



Several studies have found an association between multiple sclerosis and vitamin D (VD) deficiency, which suggests that VD may play a role in the immune response. However, few studies have addressed its role in remyelination.
DevelopmentThe VD receptor and the enzymes transforming VD into metabolites which activate the VD receptor are expressed in central nervous system (CNS) cells, which suggests a potential effect of VD on the CNS. Both in vitro and animal model studies have shown that VD may play a role in myelination by acting on factors that influence the microenvironment which promotes both proliferation and differentiation of neural stem cells into oligodendrocyte progenitor cells and oligodendrocytes. It remains unknown whether the mechanisms of internalisation of VD in the CNS are synergistic with or antagonistic to the mechanisms that facilitate the entry of VD metabolites into immune cells.
ConclusionsVD seems to play a role in the CNS and our hypothesis is that VD is involved in remyelination. Understanding the basic mechanisms of VD in myelination is necessary to manage multiple sclerosis patients with VD deficiency.
Diferentes estudios han asociado la deficiencia en VD a la esclerosis múltiple lo que ha llevado a plantear su potencial papel en la respuesta inmune. Existe menos información sobre su papel en la remielinización.
DesarrolloEn las células del SNC existe el receptor VD así como las enzimas que transforman los metabolitos de la VD para poder activar este receptor, lo que plantea un potencial efecto de la VD. Tanto estudios in vitro como modelos animales han mostrado que la VD puede tener un papel sobre la mielinización actuando en factores que influyen en el microambiente que favorece la mielinización como en la proliferación y diferenciación tanto de las células madre neuronales en células precursoras de oligodendrocitos como en éstas en oligodendrocitos. No se conoce si los mecanismos de internalización de la VD en el SNC son sinérgicos o antagónicos a los que permiten la entrada de los metabolitos de la VD en las células inmunes.
ConclusionesLa VD debe tener un papel en el SNC y se puede hipotetizar si actúa en la remielinización. El conocimiento de los mecanismos básicos de los efectos de la VD en la mielinización parece necesario para poder aconsejar a los pacientes con esclerosis múltiple ante deficiencias de VD en la clínica.
Vitamin D (VD) is a group of hormones including vitamin D2, or ergocalciferol, and vitamin D3, or cholecalciferol. VD is acquired mainly from the diet and through exposure to sunlight. Several analytical epidemiological studies have suggested an association between VD deficiency and multiple sclerosis (MS).1–6 The biological basis of this association is unknown. Hypotheses include causal mechanisms, the interaction between genetic and environmental factors, or simply the combination of multiple environmental factors. Research on the topic has focused on the role of VD and its metabolites in experimental allergic encephalomyelitis (EAE) in in vitro and animal studies.7,8 Most researchers have analysed the association between VD and the risk of inflammation, while others have evaluated the role of VD in myelination and remyelination.9 The present review addresses the latter topic.
Vitamin D deficiencyVD levels are usually determined by measuring plasma 25-hydroxyvitamin D (25[OH]D) concentration, due to the long half-life of this metabolite (15-35 days). However, the suitability of using total 25(OH)D levels to determine VD sufficiency has been questioned in recent years: detractors propose determining levels of the active forms or the free fraction, given that protein-bound metabolites may be inactive and may therefore not constitute adequate markers.10 Circulating VD may be either free or bound to albumin or to vitamin D-binding protein (DBP). The free fraction of VD constitutes a very small proportion of circulating metabolites (below 1%) and may have a different biological function from that of the protein-bound fraction.11,12 The concepts of VD sufficiency or deficiency are therefore difficult to define. Furthermore, the plasma concentration of circulating 25(OH)D varies depending on the patient's health status and certain genetic factors.13 The issue is further complicated by the fact that the free fraction of circulating VD is usually calculated through mathematical estimation rather than by direct measurement14; these 2 methods deliver considerably different results. The terminology used in the literature is also potentially confusing. The term “bioavailable” 25(OH)D is used for circulating 25(OH)D which is not bound to DBP, that is the free and albumin-bound fractions; this represents approximately 10% of total circulating 25(OH)D; this should not be mistaken for free 25(OH)D. These methodological aspects make it difficult to determine the role of VD deficiency as a risk factor for MS in general, and even more difficult in individual patients.
Plasma transport of vitamin D and transformation into active metabolitesAs mentioned previously, VD is transported to cells and tissues via transport proteins (DBP and albumin). DBP is produced in the liver. In addition to transporting VD, this protein promotes the conversion of the prohormone 25(OH)D, an inactive circulating metabolite, into the active metabolite, 1,25-dihydroxyvitamin D (1,25[OH]2D). This requires the action of 25-hydroxyvitamin D-1α-hydroxylase, also known as cytochrome p450 27B1 (CYP27B1) or simply 1-α-hydroxylase, which is encoded by the CYP27B1 gene. This enzyme is expressed in renal tubule cells and other types of cells, such as immune and central nervous system (CNS) cells. It catalyses the conversion of 25(OH)D to 1,25(OH)2D. This metabolite acts on the VD receptor (VDR), the nuclear receptor for 1,25(OH)2D. As mentioned previously, 99% of circulating 25(OH)D is bound either to DBP or to albumin,15 and only a small fraction is free.16 25(OH)D is a lipophilic molecule; therefore, it is capable of passively penetrating cell membranes. The idea that the free fraction is the active form, as occurs with other hormones, is debated for 2 main reasons. Firstly, serum concentrations of the free fraction of 1,25(OH)2D are 10−13M, nearly 1000 times lower than the concentration necessary for binding to VDR.17 Secondly, megalin, also known as low-density lipoprotein-related protein 2 (LRP2), has been shown to play an important role in VD metabolism. This transmembrane protein acts as a multiligand receptor in various tissues and binds to DBP, creating a compound that is internalised by endocytosis into the proximal tubule cells, where 1-α-hydroxylase is expressed. Megalin acts as an endocytic receptor which binds to extracellular ligands and internalises them by endocytosis, leading to the activation of certain signalling pathways involving lipoproteins, hormones, drugs, and DBP itself, which are dependent on binding with adaptor proteins that recognise specific cytoplasmic domains of megalin. These pathways are involved in protein trafficking,18 interaction with cytoskeletal or cytoplasmic proteins, and SHH signalling.19 Megalin may be phosphorylated by GSK3, PKC, CK-II, or PKA for endosomal recycling.20 As occurs with vitamin D-deficient mice, megalin-deficient mice develop the bone phenotype of rickets.21 In addition to megalin, the proteins cubilin22 and Dab2 adaptor protein23 also bind to DBP. Megalin is expressed not only in the kidney but also in the nervous system and in such other tissues as the placenta, mammary glands, and parathyroid glands. It has therefore been suggested that VD binds to megalin in these tissues.
In healthy brains, megalin is preferentially expressed in the ependymal cells lining the ventricular wall, capillaries, and choroid plexus,24 but has also been observed in the central and peripheral nervous systems. Inactivation of the megalin gene results in developmental alterations of the forebrain, absence of the olfactory apparatus, and craniofacial malformations,25 whereas a mutation in the gene results in abnormalities in the dorsal diencephalon, with hypertrophy of the choroid plexus of the third ventricle.26 In the CNS, megalin is expressed in neurons and astrocytes.27 Astrocytes need megalin to internalise albumin, which is in turn necessary for the synthesis of neurotrophic factors by nearby neurons.28 Megalin has also been found in retinal ganglion cells, where it interacts with metallothionein-IIA, enabling the activation of various intracellular signalling pathways involved in neuroprotection.29 Megalin has also been found in different neuronal populations of the cortex, hippocampus, striatum, thalamus, olfactory bulb, and cerebellum of healthy human, monkey, rat, and mouse brains. The protein is upregulated in damaged neurons in patients with Alzheimer disease.30 In the brain, megalin is involved in the endocytosis and internalisation of apolipoprotein E and the amyloid precursor protein (APP).
Internalisation of vitamin D metabolitesThe internalisation of VD metabolites is mediated by the DBP-megalin complex in various tissues. If the VD–DBP complex is present in high concentrations, more VD metabolites will be available for megalin-mediated internalisation and, consequently, the free fraction available for passive diffusion will be smaller. DBP concentration influences the availability of the free fraction of VD; an increase in the levels of circulating VD may result in a decrease in the free fraction available. On the other hand, lower DBP concentrations may increase the concentration of free VD metabolites at the cell membrane, promoting passive diffusion of these molecules. This is not always the case, however: some cells may internalise DBP by mechanisms involving proteins other than megalin, internalising bound rather than free VD metabolites. B-lymphocytes may internalise 25(OH)D by binding DBP without the presence of megalin; this may involve Fc γ receptors, which also associate with immunoglobulins. Some tissues do not express megalin, which suggests that they internalise free 25(OH)D. It has been hypothesised that free 25(OH)D especially affects immune cells (e.g. monocytes, macrophages, and dendritic cells). In vitro studies have shown that monocytes exposed to increasing doses of 25(OH)D in the presence of DBP display dose-dependent induction of antibacterial proteins. In the absence of DBP, however, this response is much more marked; the capacity of 25(OH)D to promote monocyte antibacterial activity is dependent on both serum concentration and DBP genotype.31 Similar observations have been made for regulatory T cells. However, this inverse relationship also involves other variables. Thus, VD metabolites may also bind serum albumin, although the affinity of 25(OH)D and 1,25(OH)2D for albumin is considerably lower. Furthermore, due to the relative abundance of serum albumin (650μM) compared to DBP (5μM), we should consider the possibility that VD metabolites may be transported by albumin in certain situations. As a result, part of the DBP in the serum may not be bound to any metabolite. Therefore, the binding of 25(OH)D and 1,25(OH)2D to albumin may also have an impact on the free fraction. Less than 0.1% of total circulating 25(OH)D is free. Dietary changes may decrease the binding affinity of DBP for VD metabolites; for example, polyunsaturated fatty acids can decrease this affinity, resulting in changes in the bioavailability of VD metabolites. The behaviour of free 25(OH)D in patients receiving oral and parenteral VD supplementation is unknown. However, the correlation between serum 25(OH)D and DBP concentrations seems to suggest that these supplements do not modify either total or free 25(OH)D concentrations. Total serum 25(OH)D, 1,25(OH)2D, and DBP concentrations do not change at the onset of treatment with vitamin D3 or vitamin D2 supplementation. At the end of treatment, however, the serum concentration of 25(OH)D3 increases significantly more than that of 25(OH)D2; serum DBP concentrations also change, whereas free and bioavailable 25(OH)D levels are similar in patients receiving vitamin D2 or vitamin D3 supplements. The lower efficiency of vitamin D2, compared to vitamin D3, in increasing total serum 25(OH)D levels has been attributed to the weaker affinity of 25(OH)D2 for DBP.
A third factor in the association between DBP and the free fraction is that DBP has phenotypic variations that may influence its affinity for VD metabolites. The different forms of DBP (group-specific component [Gc]1F, Gc1S, and Gc2) are associated with polymorphisms in the DBP gene or the Gc gene. These DBP variants result from changes in the protein's amino acid sequence, altering the binding affinity of DBP for VD metabolites; Gc1F displays the greatest affinity and Gc2 the lowest.32,33 This is compensated for by changes in serum DBP levels, with Gc2 levels being the highest and Gc1F levels the lowest,34 meaning that DBP concentration increases with lower affinity, and vice versa. Gc allele variability is associated with racial differences: in black and Asian populations, the Gc1F form of DBP is more prevalent, whereas Gc1S is more prevalent in white populations. Gc2, which has lower affinity for VD metabolites, is more frequent in white populations and has rarely been observed in black populations; DBP affinity is therefore linked to skin pigmentation. The phenotypic variations of DBP also include those caused by such single-nucleotide polymorphisms (SNP) as rs7041 and rs4588. These 2 SNPs are associated with lower 25(OH)D levels, and are more frequent in Hispanic and African-American populations.35 Combinations of these alleles may alter DBP concentration and affinity for 25(OH)D and 25(OH).36,37 Santos et al.38 report that the rs4588 and rs7044 SNPs are associated with lower 25(OH)D concentrations in young healthy women. Cheung et al.39 identified 4 SNPs (rs2282679, rs10741657, rs12785878, and rs6013897) linked to 25(OH)D concentration, and found that rs2282679 was associated with low serum 25(OH)D levels whereas rs12785878 was associated with VD deficiency only.
The idea that internalisation is megalin-independent in most immune cells is relevant, since it means that the internalisation of VD metabolites into these cells may be dependent on Fc γ receptors or the free fraction of VD, which has an inverse relationship with plasma DBP concentration, and consequently with plasma circulating VD concentration. This is the opposite of what occurs in CNS cells, where VD internalisation is megalin-mediated. We may therefore hypothesise that low DBP concentrations may result in a high level of internalisation into immune cells and a low level into CNS cells.
Enzymes involved in vitamin D metabolismMultiple enzymes are involved in VD metabolism.40 The first of these, CYP2R1, converts VD into 25(OH)D.41 The enzyme 1-α-hydroxylase (CYP27B1), encoded by the CYP27B1 gene, catalyses the hydroxylation of 25(OH)D into 1,25(OH)2D, which subsequently binds to VDR. The third enzyme, 1,25-dihydroxyvitamin D-24-hydroxylase (24-hydroxylase, CYP24A1), is encoded by the CYP24A1 gene. It is involved in VD degradation via 1,25(OH)2D hydroxylation, acting in the opposite direction. The 3 enzymes are members of the cytochrome P450 superfamily of enzymes. No association has been found between MS and different variants of the gene coding for DBP.42
The vitamin D receptorThe VD metabolite 1,25(OH)2D binds to VDR, a nuclear receptor. Under pathological conditions, VDR is located mainly in the cytoplasm.43 The interaction between VDR and its ligand, 1,25(OH)2D, results in 2 independent protein interaction surfaces on the VDR, promoting interaction with the retinoid X receptor (RXR), which is necessary for DNA binding, and the recruitment of co-regulators involved in gene modulation.44 After dimerising with RXR, the VDR-1,25(OH)2D complex translocates to the nucleus, where it binds to vitamin D response elements (VDRE) in VD responsive genes. Target genes may act as co-activators or co-repressors; the VDR-RXR complex induces or represses gene transcription in a process involving ATPases and proteins participating in RNA polymerase II recruitment.45 In addition to this VDRE-regulated mechanism, VDR may also inhibit genes by antagonising certain transcription factors.46,47 There are several alleles of the VDR gene. Certain variants have been associated with increased susceptibility to infections and a greater incidence of such autoimmune diseases as MS and cancer, which may be due to a decrease in VDR mRNA stability, and consequently to a decrease in VDR expression. Some variants of the VDR gene result in a non-functional receptor.48 The literature describes several polymorphisms in the gene coding for VDR (Apa-I, Taq-I, Fok-I, and Bsm-I). While some studies have found significant associations between these polymorphisms and MS,49–51 others have not.52–57
Vitamin D metabolism in the central nervous systemAlthough VD has traditionally been considered to participate in bone metabolism, its presence in the CNS suggests that it is also involved in some CNS functions; it is even considered a neurosteroid.58 A CSF study of both patients with MS and controls revealed presence of 25(OH)D, which was positively correlated with serum 25(OH)D.59 DBP was also observed in the CSF of controls, although it was more frequently found in patients with MS.60 This confirms that VD and its main transport protein enter the CNS. VDR mRNA and CYP27B1 mRNA expression in the CNS is greater in animals with EAE than in controls.61 Considering that VDR and CYP27B1 are expressed in the neurons and astrocytes of healthy individuals, it may be hypothesised that VD plays a role in certain functions of the CNS.62 In vitro studies of glial cell cultures have reported that 1,25(OH)2D3 supplementation has an anti-inflammatory effect,63 which supports the therapeutic potential of VD in MS. Considering that 1,25(OH)2D can cross the blood-brain barrier and that most CNS cells, including the microglia,64 express VDR, it seems plausible to consider that VD acts directly on the CNS. VD has recently been observed to act on histones, causing epigenetic changes65 that have been associated with MS.66,67 As mentioned previously, VDR is present in neurons and glial cells.68,69 An animal model found that 1,25(OH)2D3 increases VDR nRNA and CYP27B1 mRNA expression in female mice with EAE, compared to controls.61 CYP24A1 mRNA expression increases with exposure to 1,25(OH)2D in C6 glioma and rat primary glial cell cultures.70 IFN-γ alters CYP24A1 expression in macrophages.71 TNF and interleukin-1β production have been found to decrease in a microglial cell line exposed to 1,25(OH)2D.72 Likewise, 1,25(OH)2D3 has been shown to reduce TNF nRNA and expression of the macrophage colony-stimulating factor in primary rat astrocytes and glioma cells.73 Furthermore, MHC class II immunoreactivity decreases in EAE after treatment with 1,25(OH)2D.74
A study by Smolders et al.75 extrapolates VD metabolism in the CNS of healthy individuals to patients with MS. The researchers confirm VDR expression in the nuclei of all neurons, including in the hypothalamus. CYP24A1 is expressed in the cytoplasm of virtually all neurons, including those of the hypothalamus, and especially in the cells of the supraoptic and periventricular nuclei; in the hypothalamus, CYP24A1 co-localises with cortisol-releasing hormone, vasopressin, and oxytocin. Normal-appearing white matter of MS patients and controls shows VDR nuclear activity in oligodendrocytes. Microglia also display VDR expression, but no CYP24A expression. Astrocytes show both nuclear VDR and cytoplasmic CYP24A expression. However, some patients show cytoplasmic expression of VDR, especially in glial cells displaying increased GFAP expression. In normal-appearing white matter, glial expression of VDR mRNA and CYP27B1 mRNA is similar to that found in peripheral mononuclear cells, whereas CYP24A1 mRNA and LRP2 mRNA expression is increased in controls. VDR mRNA expression is significantly greater in patients with MS than in controls, whereas expression of CYP24A1 mRNA, CYP27B1 mRNA, and megalin mRNA is similar. The researchers observed cytoplasmic VDR expression in glial cells at the core of chronic, inactive MS lesions as well as in some cells in the perilesional white matter. VDR mRNA and CYP27B1 mRNA expression is greater in chronic, active lesions than in normal-appearing white matter. However, the researchers found no differences between chronic, inactive lesions and normal-appearing white matter. Megalin mRNA expression is lower in chronic, active and inactive lesions than in normal-appearing white matter.
The researchers include a series of in vitro experiments demonstrating the effects of VD in certain CNS cell lines. In vitro 1,25(OH)2D3 supplementation in a culture of SH-SY5Y neuroblastoma cell line, which mainly includes neurons, induces CYP24A mRNA expression.75 In vitro exposure of primary human astrocytes and astroglioma cell lines (U343 and U373) to 1,25(OH)2D3 showed dose-dependent VDR mRNA and CYP24A1 mRNA upregulation, but had no effect on CYP27B1 mRNA expression. Primary astrocyte and microglia cultures were exposed to IFN-γ, TNF, and 1,25(OH)2D3. IFN-γ and TNF were found to upregulate CYP27B1 mRNA both in astrocytes and in microglia. CYP27B1 mRNA expression was lower in cells exposed to IFN-γ or TNF and treated with 1,25(OH)2D3 than in those not treated with 1,25(OH)2D3. CYP27B1 and CYP24A1 upregulation secondary to exposure to IFN-γ/TNF and 1,25(OH)2D3 was more marked in astrocytes than in microglia. Immunohistochemical studies have found megalin in the choroidal plexus and the endothelium of brain microvessels,76 which suggests that megalin transports DBP and VD into the CNS. Furthermore, megalin mRNA expression has been observed to be lower in both active and inactive MS lesions. The mechanism by which 1,25(OH)2D promotes neuronal differentiation is unclear. It is hypothesised that the mechanism involves growth factors, based on observations that 1,25(OH)2D upregulates the expression of NT-3 and NGF,77 GDNF,78 CNTF, or BDNF.79
Vitamin D metabolism and remyelination in multiple sclerosisMyelination is the process by which axons are wrapped in myelin. Remyelination of demyelinated lesions has been observed in the early stages of MS80–82; this is supported by neuroimaging findings.83–89 However, remyelination is incomplete90 and eventually ceases,91 perhaps due to the inability of oligodendrocyte precursor cells (OPC) to migrate and reach the site of demyelination,92 or to a lack of suitable conditions for differentiation.93 Post mortem studies of patients with MS show that depletion of the OPCs available for remyelination is not a likely explanation of remyelination failure.94–97 Remyelination decreases with age, regardless of health status.98,99 The most likely explanation of this phenomenon is the inability of OPCs to mature into oligodendrocytes, probably because the microenvironment prevents OPC differentiation and subsequent axon remyelination. This is relevant for MS treatment since current treatments are effective only in controlling immune mechanisms (that is, in the early stages of the disease) but have no effect on remyelination.
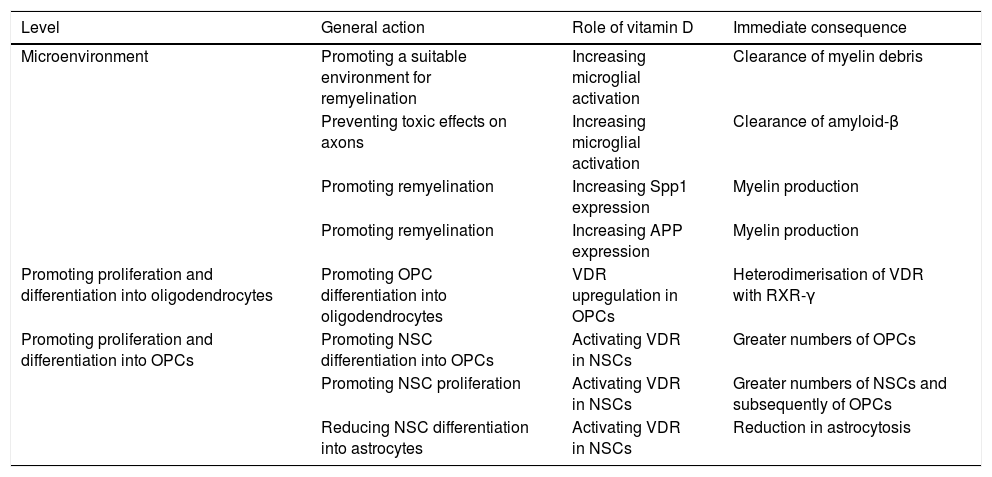
Little is known about the role of VD in myelination. According to a number of studies, however, VD may be involved in myelination and remyelination at different levels (Table 1).100–108 Combined treatment with myelin peptides and VD protects against EAE103,104 and an animal model of Krabbe disease.105 Given that VD decreases inducible nitric acid synthase expression in the microglia, it may have an impact on the balance between the inflammatory and anti-inflammatory mechanisms involved in remyelination. VD also increases microglial activation, promoting the clearance of myelin debris and, as a consequence, remyelination.106 In OPC cell cultures, VD upregulates the transcription of VDR and NGF mRNA, but not of myelin basic protein or proteolipid protein mRNA. As mentioned previously, oligodendrocytes express VDR, and 1,25(OH)2D depletion results in reduced differentiation into oligodendrocytes and demyelination107; VD and VDR therefore positively regulate OPC differentiation. VDR expression has been observed in OPCs in MS tissue cultures.108,109 In OPCs, VDR heterodimerises with RXR-γ and participates in OPC differentiation; RXR-γ is expressed in OPCs during remyelination.110 In vitro studies show that blocking VDR reduces OPC differentiation, myelination, and remyelination, whereas activating VDR via VD increases differentiation.111 Likewise, neural stem cells (NSC) express VDR and 1,25(OH)2D. The latter increases NSC proliferation and differentiation into neurons and oligodendrocytes, reducing astrogliosis.112 VD promotes NSC proliferation.113 It has been suggested that Spp1, a VD-regulated cytokine, plays a role in MS.114 Spp1 boosts myelin formation in vitro115; and its expression has been shown to increase during remyelination in an animal model of toxin-induced demyelination.116
Potential roles of vitamin D at different levels of the remyelination process.
| Level | General action | Role of vitamin D | Immediate consequence |
|---|---|---|---|
| Microenvironment | Promoting a suitable environment for remyelination | Increasing microglial activation | Clearance of myelin debris |
| Preventing toxic effects on axons | Increasing microglial activation | Clearance of amyloid-β | |
| Promoting remyelination | Increasing Spp1 expression | Myelin production | |
| Promoting remyelination | Increasing APP expression | Myelin production | |
| Promoting proliferation and differentiation into oligodendrocytes | Promoting OPC differentiation into oligodendrocytes | VDR upregulation in OPCs | Heterodimerisation of VDR with RXR-γ |
| Promoting proliferation and differentiation into OPCs | Promoting NSC differentiation into OPCs | Activating VDR in NSCs | Greater numbers of OPCs |
| Promoting NSC proliferation | Activating VDR in NSCs | Greater numbers of NSCs and subsequently of OPCs | |
| Reducing NSC differentiation into astrocytes | Activating VDR in NSCs | Reduction in astrocytosis |
VD-induced microglial activation promotes phagocytosis of amyloid-β peptides (Aβ),117 thus preventing axon damage: Aβ expression is increased in demyelinating plaques,118–120 which, although initially beneficial, may cause toxicity at later stages. Megalin is involved in APP and Aβ endocytosis and internalisation.121 A megalin-APP-Fe65 complex has been found in hippocampal neurons; this complex would act as a regulator of Aβ toxicity.122 We have recently studied the potential role of APP and its signalling pathway123 based on PET imaging findings of amyloid tracer uptake in myelin.124 APP is upregulated in demyelinated axons, which activates the amyloid cascade, promoting Aβ production.125,126 APP expression also increases after spinal white matter compression injury in rats.127 VD increases APP expression in rats128 and is thought to play a role in remyelination.
ConclusionsIn advanced stages of MS, the ability to remyelinate axons is lost.81,129 This is relevant to MS treatment: current treatments control immune mechanisms and are therefore effective only in the early stages of the disease; they have no effect on remyelination and consequently on sequelae.130,131 VD metabolism is present in the CNS, participates in myelination, and may be influenced by such external factors as diet, sun exposure, or VD supplementation. Understanding the role of VD in myelination may eventually allow us to better manage patients with MS and VD deficiency.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Matías-Guíu J, Oreja-Guevara C, Matias-Guiu JA, Gomez-Pinedo U. Vitamina D y remielinización en la esclerosis múltiple. Neurología. 2018;33:177–186.
