





La adrenomieloneuropatía es una de las manifestaciones de adrenoleucodistrofia-X, ubicada dentro de los 6 fenotipos de este trastorno neurodegenerativo.
Caso clínicoVarón de 42 años inició la sintomatología hace 22 años, caracterizada por paraparesia progresiva y dificultad en la marcha, continuando en la adultez con hiperpigmentación gingival y mamilar, paraparesia espástica e hiperreflexia. La resonancia magnética de columna vertebral mostró alteración a nivel de T2-T7, estableciendo el diagnóstico mediante valores plasmáticos superiores de ácidos grasos de cadena muy larga.
ConclusionesSe reporta un caso de adrenomieloneuropatía en el Perú que cumple con los criterios clínicos, imagenológicos y laboratoriales.
Adrenomyeloneuropathy is one of the manifestations of adrenoleukodystrophy-X, which is found within the 6 phenotypes of this neurodegenerative disorder.
Case reportA 42-year-old male started the symptoms 22 years ago, characterized by a progressive paraparesis and difficulty in march, continuing in adulthood with gingival and mamilar hyperpigmentation, spastic paraparesis and hyperreflexia. The magnetic resonance of the spine showed alteration at the level of T2-T7, establishing the diagnosis by higher plasmatic values of very long chain fatty acids.
ConclusionsA case of adrenomyeloneuropathy is reported in Peru that meets the clinical, imaging and laboratory criteria.
La adrenomieloneuropatía (AMN) es un fenotipo de presentación de la adrenoleucodistrofia relacionada con el cromosoma X (ALD-X)1 caracterizado por una alteración en el catabolismo de los ácidos grasos de cadena muy larga (AGCML) por un defecto en la beta-oxidación peroxisomal que provoca una acumulación anormal de AGCML en los tejidos y fluidos corporales; afectando principalmente a la corteza suprarrenal, la sustancia blanca del sistema nervioso y los testículos2.
Las lesiones de AMN se producen principalmente en la médula espinal y los nervios periféricos, y se manifiestan con clínica de paraparesia espástica lentamente progresiva y disfunciones sensoriales y autonómicas relacionadas a una degeneración axonal posterior de los tractos corticoespinales y dorsales3. Los pacientes con AMN suelen mostrar en la resonancia magnética una atrofia difusa en la médula espinal, principalmente en las regiones torácicas (90%)4, y en la resonancia magnética del cerebro muestran infrecuentemente desmielinización de la sustancia blanca, siendo la mayoría hallazgos de normalidad. La espectroscopía es la prueba que distingue entre la ALD y la AMN «puro» y en ella se observan cambios axonales5.
La concentración plasmática de AGCML está elevada en casi el 100% de los varones y en aproximadamente el 85% de las mujeres portadoras, por ello en pacientes masculinos el aumento en los niveles plasmáticos de AGCML o la mutación patogénica en el gen ABCD1 corrobora el diagnóstico6.
Para definir el cuadro clínico del paciente se aplicaron criterios diagnósticos de AMN (tabla 1). A continuación, presentamos un caso clínico y una breve revisión de la literatura.
Criterios diagnósticos
| - Estudio de la mutación patogénica del gen ABCD1 |
| - Valores incrementados de AGCML en el plasma |
| - Antecedentes familiares de ALD y sus variantes |
| - Cuadro clínico caracterizado por paraparesia flácida progresiva e insuficiencia suprarrenal (enfermedad de Addison) |
| - La RMN encefálica no presentó alteraciones y la RMN de columna vertebral mostró un adelgazamiento en la región dorsal de la médula espinal |
| - Estudios neurofisiológicos con polineuropatía desmielinizante sensitivo motora de grado moderado |
AGCML: ácidos grasos de cadena muy larga; ALD: adrenoleucodistrofia; RMN: resonancia magnética nuclear.
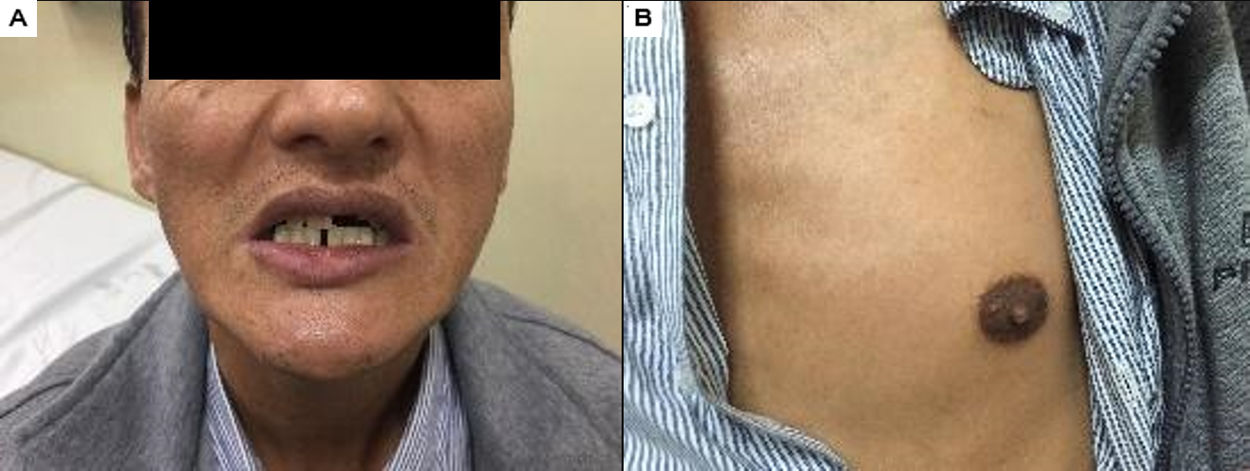
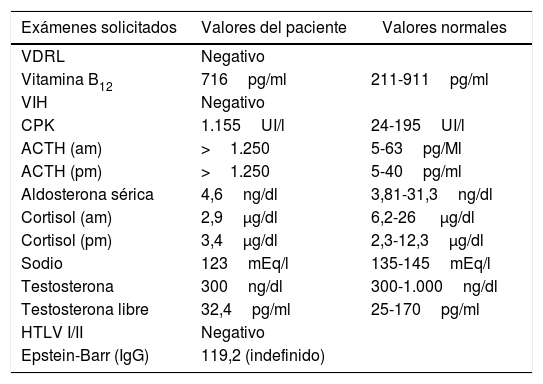
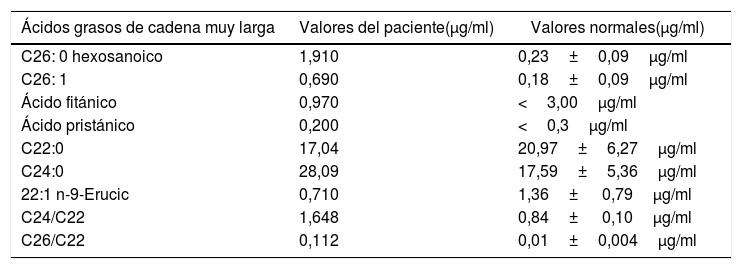
El caso reportado corresponde a un varón de 42 años de Pozuzo, Oxapampa, Perú, con familiares con manifestaciones clínicas de adrenoleucodistrofia, aunque sin confirmación genética o acumulación de AGCML, que comenzó sintomatología de forma insidiosa hace 22 años caracterizada por una disminución progresiva de la fuerza en miembros inferiores, dificultad en la marcha sin alteración sensitiva, esfinteriana ni marcha atáxica. Ingresó por el servicio de urgencia en un hospital estatal en Perú en mayo de 2015 debido a náuseas, vómitos, hipotensión, convulsiones, fatiga, hiporexia y pérdida de peso. En el examen físico presentó hiperpigmentación gingival y mamaria (fig. 1), paraparesia espástica, hiperreflexia (MRSS: 4/5), Ashworth 2 y Babinski bilateral. Los resultados de las pruebas de laboratorio mostraron un aumento de CPK y ACTH, y una disminución de cortisol (am) y sodio sérico: 123 mEq/l (tabla 2). Asimismo, los valores de AGCML hallados fueron elevados (tabla 3).
 y mamilar (B).")
Resultados de prueba de laboratorio
| Exámenes solicitados | Valores del paciente | Valores normales |
|---|---|---|
| VDRL | Negativo | |
| Vitamina B12 | 716pg/ml | 211-911pg/ml |
| VIH | Negativo | |
| CPK | 1.155UI/l | 24-195UI/l |
| ACTH (am) | >1.250 | 5-63pg/Ml |
| ACTH (pm) | >1.250 | 5-40pg/ml |
| Aldosterona sérica | 4,6ng/dl | 3,81-31,3ng/dl |
| Cortisol (am) | 2,9μg/dl | 6,2-26 μg/dl |
| Cortisol (pm) | 3,4μg/dl | 2,3-12,3μg/dl |
| Sodio | 123mEq/l | 135-145mEq/l |
| Testosterona | 300ng/dl | 300-1.000ng/dl |
| Testosterona libre | 32,4pg/ml | 25-170pg/ml |
| HTLV I/II | Negativo | |
| Epstein-Barr (IgG) | 119,2 (indefinido) |
ACTH: hormona adrenocorticotrópica; CPK: creatina-fosfocinasa; HTLV: virus linfotrópico humano de células T; VDRL: Venereal Disease Research Laboratory; VIH: virus de inmunodeficiencia humana.
Resultados del estudio de ácidos grasos de cadena muy larga
| Ácidos grasos de cadena muy larga | Valores del paciente(μg/ml) | Valores normales(μg/ml) |
|---|---|---|
| C26: 0 hexosanoico | 1,910 | 0,23±0,09μg/ml |
| C26: 1 | 0,690 | 0,18±0,09μg/ml |
| Ácido fitánico | 0,970 | <3,00μg/ml |
| Ácido pristánico | 0,200 | <0,3μg/ml |
| C22:0 | 17,04 | 20,97±6,27μg/ml |
| C24:0 | 28,09 | 17,59±5,36μg/ml |
| 22:1 n-9-Erucic | 0,710 | 1,36± 0,79μg/ml |
| C24/C22 | 1,648 | 0,84± 0,10μg/ml |
| C26/C22 | 0,112 | 0,01±0,004μg/ml |
Realizado en el Laboratorio de Genética del Instituto Kennedy-Krieger, Baltimore, EE. UU.
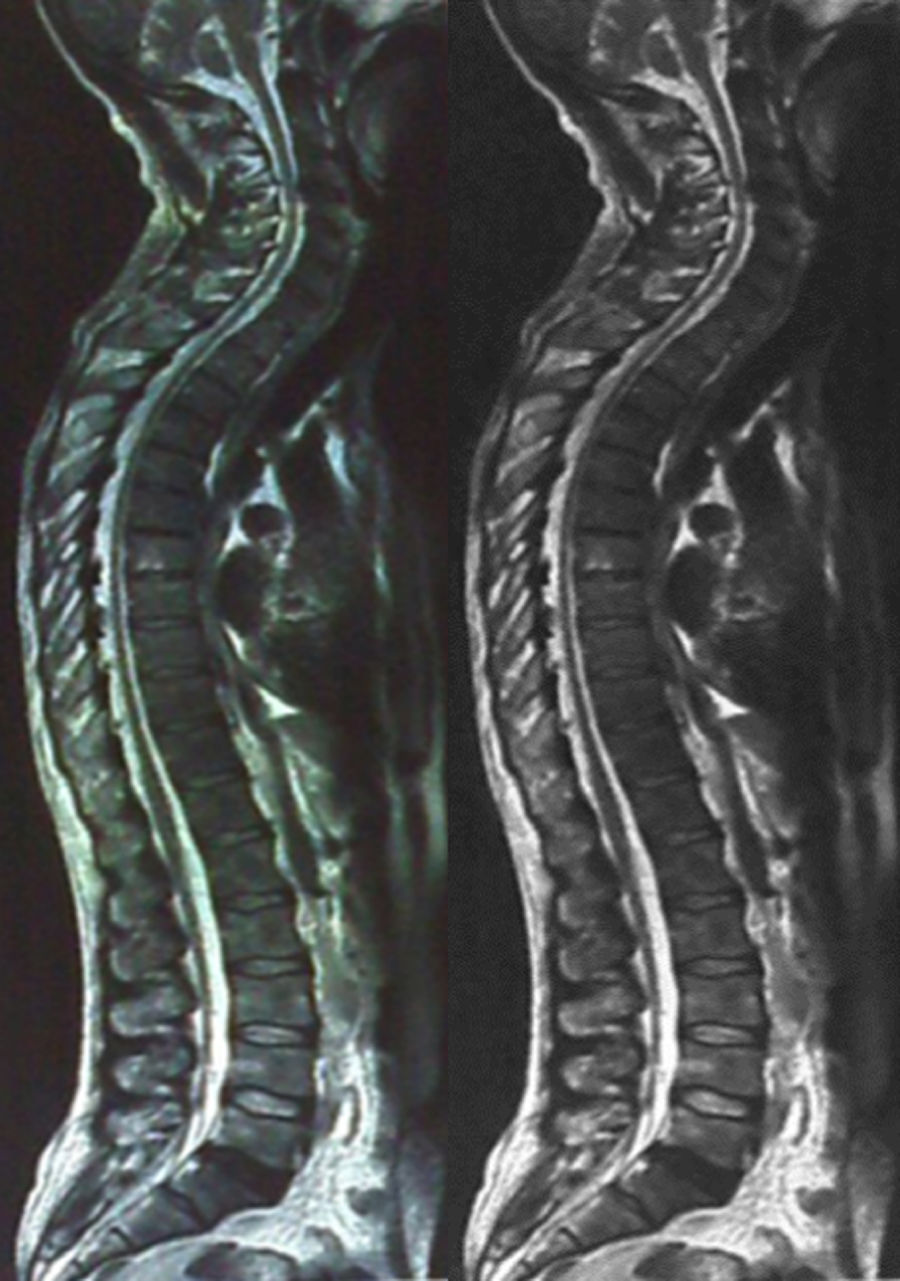
La resonancia magnética del cerebro no mostró alteraciones; sin embargo, la médula espinal en su vista panorámica mostró adelgazamiento marcado a nivel torácico de T2 a T7 (fig. 2). El estudio de la electromiografía y la velocidad de conducción nerviosa mostraron alteraciones compatibles con polineuropatía desmielinizante sensitivo motora de grado moderado.

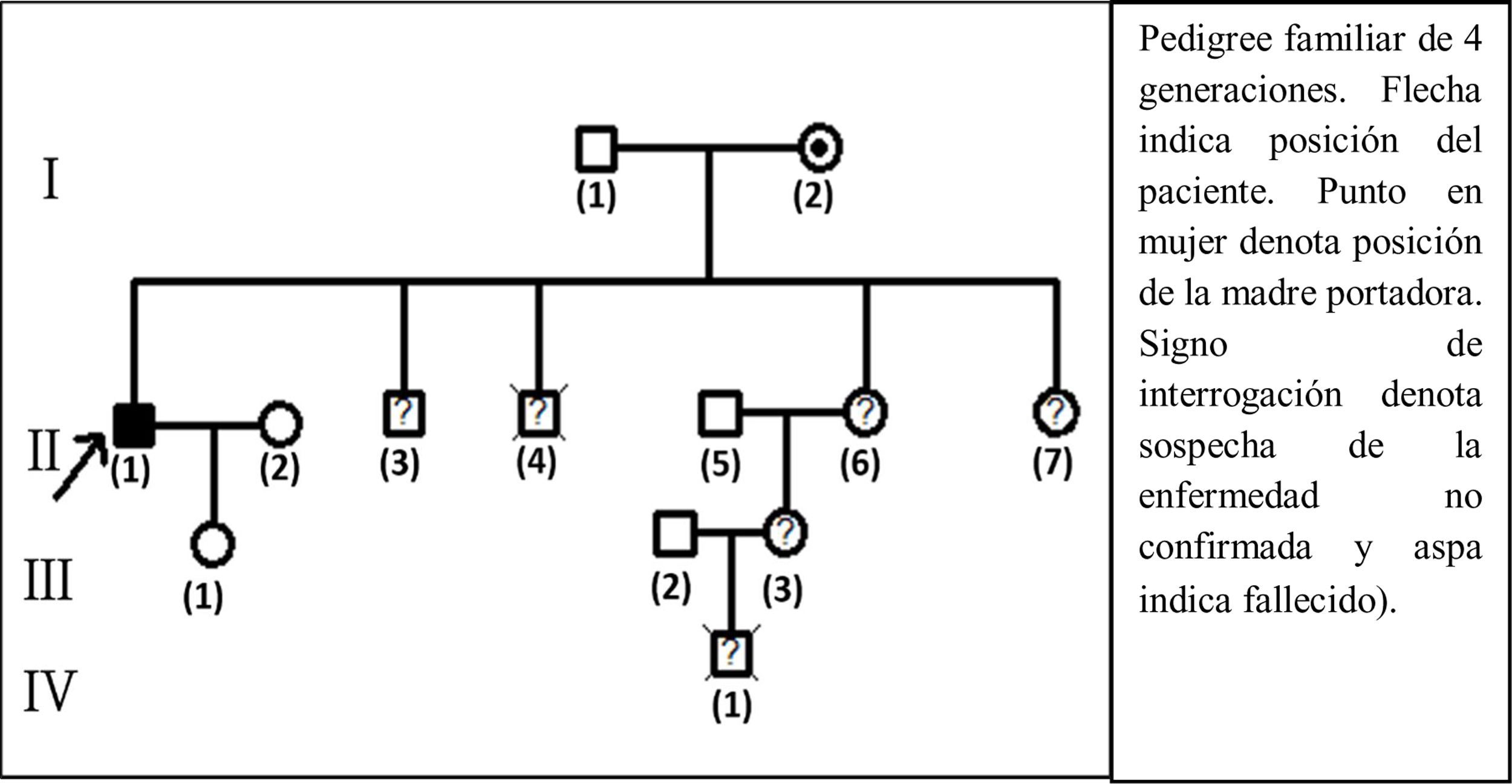
Como se muestra en la figura 3, el árbol genealógico muestra un patrón de herencia recesiva ligada al cromosoma X, donde se observa la afectación familiar.
 madre portadora; II (1) paciente caso; II (3) hermano con síntomas similares durante la segunda década de vida; II (4) hermano fallecido a los 7 años; II (6), II (7) hermanas y III (3) sobrina con sospecha de la enfermedad; IV (1) sobrino nieto que falleció a los 7 años habiendo presentado retraso del desarrollo psicomotor.")
Pedigree familiar de 4 generaciones: I (2) madre portadora; II (1) paciente caso; II (3) hermano con síntomas similares durante la segunda década de vida; II (4) hermano fallecido a los 7 años; II (6), II (7) hermanas y III (3) sobrina con sospecha de la enfermedad; IV (1) sobrino nieto que falleció a los 7 años habiendo presentado retraso del desarrollo psicomotor.
La AMN es una forma de ALD de aparición tardía que afecta a adultos entre 20 y 40 años7. En la mayoría de los casos, los pacientes con AMN no tienen compromiso cerebral clínicamente significativo, y solo el 20% de los varones puede tener evidencia de desmielinización cerebral en resonancia magnética8. Los pacientes desarrollan gradualmente paraparesia espástica progresiva, ataxia sensorial con sensación alterada de vibración, disfunción del esfínter (principalmente urinaria), dolor en las piernas y disfunción sexual9.
Las lesiones desmielinizantes, tanto periféricas como centrales, son heterogéneas y estarían involucradas en las manifestaciones clínicas de AMN, aunque se desconocen los mecanismos fisiopatológicos exactos, podrían depender de los anticuerpos de las estructuras neuronales10. Básicamente, se presenta una axonopatía distal y la respuesta inflamatoria es inexistente o muy leve. La lesión espinal consiste en la pérdida axonal y de mielina principalmente en los tractos grácil y corticoespinal. Aparentemente no hay pérdida neuronal, sin embargo, se han observado numerosas inclusiones de lípidos mitocondriales en las neuronas en casos de AMN11.
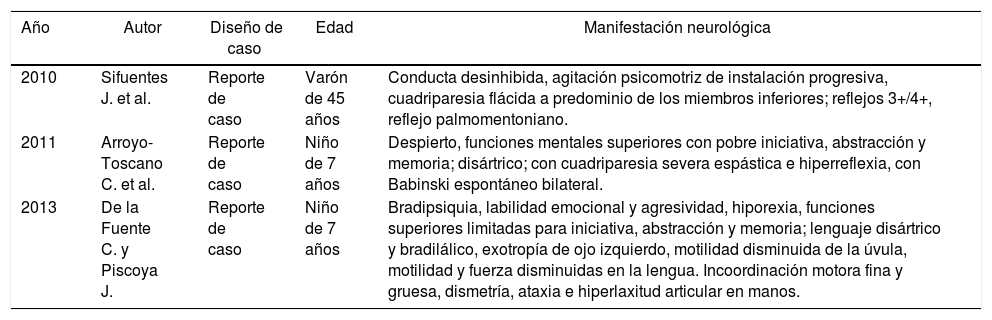
En Perú los informes son escasos, como se muestra en la tabla 4, se identificaron 3 casos de pacientes con ALD en los últimos 10 años, con síntomas neurológicos diferentes a los de nuestro paciente, que desarrollaron la enfermedad por estar ligada al cromosoma X, siendo 2 niños de 7 años12,13 y un adulto de 45 años14.
Reportes de caso en Perú
| Año | Autor | Diseño de caso | Edad | Manifestación neurológica |
|---|---|---|---|---|
| 2010 | Sifuentes J. et al. | Reporte de caso | Varón de 45 años | Conducta desinhibida, agitación psicomotriz de instalación progresiva, cuadriparesia flácida a predominio de los miembros inferiores; reflejos 3+/4+, reflejo palmomentoniano. |
| 2011 | Arroyo-Toscano C. et al. | Reporte de caso | Niño de 7 años | Despierto, funciones mentales superiores con pobre iniciativa, abstracción y memoria; disártrico; con cuadriparesia severa espástica e hiperreflexia, con Babinski espontáneo bilateral. |
| 2013 | De la Fuente C. y Piscoya J. | Reporte de caso | Niño de 7 años | Bradipsiquia, labilidad emocional y agresividad, hiporexia, funciones superiores limitadas para iniciativa, abstracción y memoria; lenguaje disártrico y bradilálico, exotropía de ojo izquierdo, motilidad disminuida de la úvula, motilidad y fuerza disminuidas en la lengua. Incoordinación motora fina y gruesa, dismetría, ataxia e hiperlaxitud articular en manos. |
En la mayoría de los pacientes, la combinación de características clínicas, antecedentes familiares de ALD, alteración de los estudios neurofisiológicos y los resultados de imágenes son causa de sospecha clínica, confirmándose los criterios diagnósticos a través de estudios genéticos y niveles elevados de AGCML. Como se muestra en la figura 3, en la familia del paciente, los varones son sospechosos de presentar la enfermedad debido a sus manifestaciones clínicas, mientras que las mujeres son posiblemente portadoras. Esto se explica por qué la AMN es una enfermedad ligada al cromosoma X, en la cual el gen anormal puede manifestarse tanto a edades tempranas como en adultos.
Para determinar el tipo de asesoramiento genético, es importante reconocer que los varones fallecidos tempranamente pueden haber desarrollado las formas más agresivas de la enfermedad ALD, a diferencia de las mujeres que son asintomáticas, pero que la identificación de su estado de portadoras podría tener un valor agregado en la prevención de nuevos casos15.
Otros casos notificados en todo el mundo de AMN fueron: un varón de 16 años16; personas entre 29 y 32 años17,18 y un varón adulto intermedio de 45 años19. Presentaron manifestaciones clínicas como debilidad de miembros inferiores de evolución lenta, con paraparesia espástica y ataxia; y todos ellos con alteración en la cantidad de AGCML. El primer caso fue peculiar porque las manifestaciones clínicas aparecieron durante la primera década de la vida, a diferencia de los casos restantes, donde el inicio de los síntomas fue durante la tercera y cuarta década de la vida.
Asimismo, se reportaron series de casos en otros países como Brasil, el cual consistía en 24 casos de ALD, 23 de ellos varones, entre los cuales había 19 casos de ALD cerebral, 3 casos de AMN y un solo caso de solamente afectación suprarrenal20. Esta situación se asemejaría a la vista en Perú, donde, según la revisión bibliográfica realizada, no se encontraron afectaciones solamente suprarrenales. Otra serie de casos realizada en China reportaba 6 casos de AMN con inicio de síntomas entre los 21 y 28 años, lo cual se asemeja a nuestro caso reportado, con compromiso espinal y solo 2 casos con afectación cerebral, a diferencia de los casos en Perú donde el compromiso solo se reporta a nivel espinal del SNC21. De igual forma, en esta serie de casos en China, todos los pacientes presentaron paraparesia espástica y cambios de coloración en la piel o cabello, similar con lo reportado en nuestro caso.
El realizar un seguimiento a los familiares de un paciente ya diagnosticado con ALD puede develar muchos casos más debido a su componente hereditario ligado al cromosoma X. Esto puede comprobarse en una serie de casos dentro de una familia en Irán22, donde se hizo un seguimiento a los demás miembros de una familia cuyos antepasados ya habían presentado sintomatologías sugerentes de ALD, encontrándose 5 varones y una mujer con sintomatología de ALD.
Por lo tanto, en pacientes con debilidad muscular de miembros inferiores con progresión lenta y adelgazamiento de la médula espinal, es importante considerar la AMN como uno de los diagnósticos diferenciales. No se han identificados otros reportes de caso en Perú con características clínicas del fenotipo típico de AMN sin afectación cerebral, confirmado por estudios genéticos o de laboratorio, en quien también se ha llevado a cabo un estudio de la genealogía de su familia, identificando un patrón de herencia vinculado al cromosoma X, donde los descendientes varones estuvieron afectados y las mujeres posiblemente fueron portadoras.
El tratamiento está dirigido a controlar complicaciones como la insuficiencia suprarrenal mediante el reemplazo con corticoides, el trasplante de células hematopoyéticas para los fenotipos de inicio temprano (cerebral infantil), la terapia nutricional con restricción dietética y el uso de estatinas como la lovastatina además de fenilbutirato para reducir los AGCML. Los resultados de la terapia génica aún son preliminares y aún no han mostrado beneficios claros23.
FinanciaciónLos autores no han declarado fuente alguna de financiamiento para este artículo.
Conflicto de interesesLos autores no informan sobre conflictos de intereses en este artículo.
