






La distrofia muscular de Duchenne (OMIM: 310200, ORPHA: 98896) es una miopatía hereditaria, recesiva, ligada a X. Es el desorden neuromuscular de etiología genética más frecuente, con una incidencia de uno por cada 3.500 hombres y una prevalencia global de 4,78 por cada 100.000 personas.
MétodosSe llevó a cabo una evaluación clínica de 63 pacientes con distrofia muscular de Duchenne en un centro de referencia de Colombia. Se realizó a todos los sujetos un análisis molecular a través de MLPA y una secuenciación masiva en paralelo.
ResultadosEn 47 de los 62 sujetos (75%) se detectaron variantes tipo deleción/duplicación de exones en el gen de la distrofina, mediante MLPA. Se encontraron 33 probandos con deleciones (53,2%) y 14 casos de duplicaciones (22,5%) de uno o más exones. El estudio de secuenciación del gen DMD por la técnica de secuenciación masiva en paralelo fue realizado en 15 individuos (24,2%) con MLPA negativo. Se evidenciaron mutaciones sin sentido (nonsense) en 6 casos (9,6%), mutaciones que afectan el marco de lectura en 7 casos (11%) y mutaciones que afectan el splicing en 2 probandos (3,2%). No se encontraron mutaciones missense. Las variantes se distribuyeron a lo largo del gen y se encontraron 5 variantes que no habían sido previamente reportadas, identificadas en 7 individuos.
ConclusionesEl diagnóstico molecular de mutaciones puntuales a través de secuenciación masiva paralela permitió alcanzar de manera eficiente la confirmación diagnóstica, definiendo para los casos negativos para deleción/duplicación, la asesoría adecuada y la implementación de un manejo terapéutico apropiado.
Muscular dystrophy of Duchenne (OMIM: 310200, ORPHA: 98896) is a recessive X-linked primary myopathy. It is the most frequent neuromuscular disorder of genetic etiology, with an incidence of one per 3,500 men and an overall prevalence of 4.78 per 100.000 people.
MethodsA clinical evaluation of 63 patients with Duchenne muscular dystrophy was carried out in a Colombian reference center. We performed all the subjects, molecular analysis through MLPA and massive sequencing in parallel.
ResultsIn 47 of the 62 subjects (75%), deletion/duplication variants of exons were detected in the dystrophin gene, using MLPA. We found 33 probands with deletions (53.2%) and 14 cases of duplications (22.5%) of one or more exons. The sequencing study of the DMD gene by the mass sequencing technique in parallel was performed in 15 individuals (24.2%) with negative MLPA. There were nonsense mutations in 6 cases (9.6%), mutations affecting the reading frame in 7 cases (11%) and mutations affecting splicing in 2 cases (3.2%). No missense mutations were found. The variants were distributed throughout the gene, and five variants were found that had not been previously reported, identified in 7 individuals.
ConclusionsThe molecular diagnosis of point mutations through massively parallel sequencing allowed to reach the diagnostic confirmation efficiently, defining for the negative cases for deletion/duplication, the adequate advice and the implementation of appropriate therapeutic management.
Las miopatías son un grupo heterogéneo de enfermedades que afectan el músculo; se clasifican en miopatías primarias (distrofias musculares, miopatías congénitas y miopatías metabólicas) y miopatías adquiridas (de origen endocrinológico, metabólico, reumatológico, inflamatorio o secundario al uso de medicamentos)1. Las distrofias musculares más frecuentes son las de origen genético y se caracterizan por degeneración y debilitamiento progresivo, principalmente del músculo esquelético2. Las mutaciones que originan estos trastornos afectan proteínas de la membrana plasmática, de la matriz extracelular o proteínas de la membrana nuclear, alterando la integridad de la célula y generando ciclos sucesivos de degeneración y regeneración de la fibra muscular, con consecuencias deletéreas, como la atrofia muscular y el posterior reemplazo de la fibra por tejido graso y fibroso3.
La distrofia muscular de Duchenne (DMD, OMIM: 310200, ORPHA: 98896) es una miopatía hereditaria, recesiva, ligada a X. Es el desorden neuromuscular de etiología genética más frecuente, con una incidencia de uno por cada 3.500 hombres y una prevalencia global de 4,78 por cada 100.0004,5. Tiene un espectro de severidad variable, que incluye desde una presentación muy severa, como la DMD, hasta una presentación atenuada, como es el caso de la distrofia muscular de Becker (DMB). La forma severa se caracteriza por un desgaste muscular proximal progresivo, que comienza alrededor de los 3 años de edad y se caracteriza clínicamente por retraso en el desarrollo motor, debilidad proximal, pseudohipertrofia de los gastrocnemios, elevación en los niveles de creatina cinasa (CK) en sangre y cambios miopáticos en la electromiografía. Alrededor de los 12 años de edad, el paciente requiere el uso de silla de ruedas para su movilización. La enfermedad culmina con insuficiencia respiratoria, falla cardiaca y muerte prematura alrededor de los 20 años de edad del sujeto afectado3,5. En el caso de la DMB, esta inicia sus manifestaciones entre los 20 y los 30 años de edad, con compromiso leve del músculo esquelético, y su morbilidad más relevante es la falla cardiaca y las arritmias asociadas6; las arritmias tienen una amplia variedad de presentación clínica, que incluye: falla cardiaca en el adulto joven, muerte súbita cardiaca o incluso anormalidades subclínicas7.
El gen causante de la DMD/DMB (Xp21.2) es uno de los más largos del genoma humano (≈2,4Mb) y está compuesto por 79 exones8. El gen DMD (Xp21.2-p21.1) codifica la proteína intracelular distrofina, que participa dentro de un complejo de proteínas relacionado con la resistencia al estrés mecánico, generado durante la contracción muscular9. Se han descrito diversas formas de mutaciones en el gen DMD; entre el 65 y el 70% de los casos se debe a deleciones de uno o más exones, mientras que las duplicaciones comprenden entre el 7 y el 18%. El resto de los casos se deben a mutaciones puntuales10,11.
Actualmente solo se dispone de manejo paliativo para la DMD, el cual se basa en terapia física, el uso de apoyos ortopédicos y el tratamiento de las complicaciones respiratorias y cardiacas, junto con la administración de glucocorticoides12. Algunos estudios clínicos han indicado que los glucocorticoides pueden prolongar la marcha y mejorar la función y la fortaleza muscular13,14. Sin embargo, generan varios efectos adversos, sin modificar la función de la proteína distrofina y, por ende, sin impactar significativamente sobre la etiología de la enfermedad y su pronóstico a largo plazo14. En este contexto, han surgido terapias potenciales para la DMD, siendo las más prometedoras los oligonucleótidos antisentido que inducen salto del exón (exon skipping)15–17 y las terapias de salto de codón de parada temprano originadas en mutaciones sin sentido18. De tal modo que ante la especificidad de los tratamientos es necesaria la caracterización molecular de la enfermedad, tanto para la consejería genética como para evaluar el uso de manejo farmacológico19. Por otra parte, la caracterización por grupos poblacionales permite establecer prevalencias globales y comparar las frecuencias de variantes genéticas entre grupos humanos20.
El presente estudio recoge el espectro mutacional de un grupo de pacientes colombianos con diagnóstico de DMD, contribuyendo a la caracterización molecular de la DMD/DMB y ampliando el conocimiento sobre el espectro y las frecuencias de mutaciones de la enfermedad.
Población y métodosEl presente estudio fue aprobado por el Comité de Ética de la Investigación de la Facultad de Medicina de la Pontificia Universidad Javeriana, de Bogotá D. C. (Colombia) y del Hospital Universitario San Ignacio. Todos los sujetos se sometieron al proceso de consentimiento informado y asentimiento informado de los menores de 18 años.
PoblaciónSe incluyeron 62 sujetos masculinos remitidos al Instituto de Genética Humana (Facultad de Medicina de la Pontificia Universidad Javeriana) por sospecha clínica de DMD. Los motivos de sospecha clínica fueron debilidad muscular progresiva, niveles elevados de CK, hipertrofia de gastrocnemios, signo de Gowers positivo, cambios miopáticos en la electromiografía y antecedentes familiares. Después de proveer el consentimiento informado, se extrajo a cada paciente ADN de leucocitos a partir de una muestra de sangre periférica utilizando el método de extracción Salting Out según lo descrito por Miller et al.21. Para cada sujeto se realizó análisis por amplificación de sondas por ligación múltiple (MLPA) para deleciones y duplicaciones del gen de la distrofina. En los casos en los que el MLPA no detectó deleciones o duplicaciones, se procedió a realizar la secuenciación del gen.
Análisis por amplificación de sondas dependiente de ligandos múltiplesLa técnica de MLPA se llevó a cabo de acuerdo con las instrucciones del fabricante, utilizando el kit SALSA MLPA P034 y P035 (MRC-Holland, Amsterdam, Países Bajos). El kit SALSA P034 contiene sondas para los exones 1-10, 21-30, 41-50 y 61-70 del gen de la distrofina, mientras que el kit SALSA P035 contiene sondas para los exones 11-20, 31-40, 51-60 y 71-79. Inicialmente, 15ng de ADN fueron denaturados y durante la noche se hibridaron las sondas de la mezcla P034 y P035. Posteriormente, con la enzima ligasa 65, se llevó a cabo el proceso de ligamiento de las sondas y, finalmente, la reacción en cadena de la polimerasa (PCR) utilizando los primers específicos SALSA FAM PCR primers. Los productos amplificados y el estándar de tamaño se mezclaron y se corrieron en un sistema de análisis genético ABI 3100 (Applied Biosystems, Thermo Fisher Scientific, Waltham, Massachusetts, EE. UU.). El análisis se realizó por medio de Coffalyser.Net software (MRC-Holland, Amsterdam, Países Bajos).
Análisis por secuenciación masiva en paraleloEl ADN genómico fue enriquecido para las regiones seleccionadas (coordenadas genómicas ChrX:31 047 266 a 33 139 594 NM_004006, hg19) utilizando un protocolo basado en hibridación, y se secuenció utilizando la tecnología Illumina®. Todas las regiones seleccionadas tenían una profundidad mayor o igual a 50× y un umbral mínimo de confianza de mapeo de Q30. Las lecturas fueron alineadas contra la secuencia de referencia (GRCh37) y los cambios fueron identificados e interpretados en el contexto de un único transcrito clínicamente relevante. Todas las variantes patogénicas y probablemente patogénicas fueron confirmadas por secuenciación de Sanger. La clasificación de las variantes se realizó de acuerdo con los criterios de la ACMG22 y se utilizaron como referencia las bases de datos Leiden Open Variation Database23, UMD-DMD24 y Proyecto 1000 genomas25. Para el análisis in silico de la patogenicidad de las mutaciones, se aplicaron los algoritmos SIFT26, PolyPhen-227 y Align-GVGD28,29.
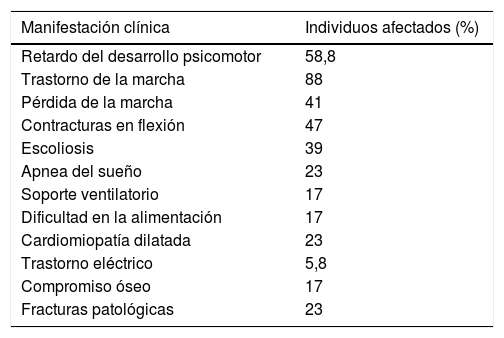
ResultadosUn total de 62 pacientes con sospecha clínica de DMD/DMB fueron confirmados por análisis molecular (MLPA o secuenciación). La edad promedio de valoración de los individuos fue de 9 años, con un rango de 8 meses a 27 años. La edad de comienzo de los síntomas fue menor de 4 años en el 79% de los pacientes. Los síntomas iniciales de la enfermedad fueron: caídas frecuentes, debilidad muscular progresiva, dificultad para levantarse, dificultad para subir escaleras, dolor muscular y retardo del neurodesarrollo asociado a niveles elevados de CK (tabla 1). En el momento del estudio 22 pacientes (35,4%) habían perdido la marcha, el más temprano a la edad de 6 años, con un promedio de pérdida de marcha, para todo el grupo, de 9 años. El compromiso cardiaco (fracción de eyección inferior al 55%) se desarrolló a una edad media de 14±3,1 años. Ninguno recibió trasplante cardiaco. El 33% de los probandos reportó algún antecedente familiar de enfermedad neuromuscular, lo que incluyó hermanos y primos. En relación con casos familiares, se evaluaron en el estudio 7 parejas de hermanos, un par de primos hermanos de una sola familia y 2 hermanos y un primo de otra familia.
Manifestaciones clínicas de pacientes con distrofia muscular de Duchenne (n=63)
| Manifestación clínica | Individuos afectados (%) |
|---|---|
| Retardo del desarrollo psicomotor | 58,8 |
| Trastorno de la marcha | 88 |
| Pérdida de la marcha | 41 |
| Contracturas en flexión | 47 |
| Escoliosis | 39 |
| Apnea del sueño | 23 |
| Soporte ventilatorio | 17 |
| Dificultad en la alimentación | 17 |
| Cardiomiopatía dilatada | 23 |
| Trastorno eléctrico | 5,8 |
| Compromiso óseo | 17 |
| Fracturas patológicas | 23 |
En 47 de los 62 sujetos (75%) se detectaron variantes tipo deleción/duplicación de exones en el gen de la distrofina, mediante MLPA. Se encontraron 33 probandos con deleciones (53,2%) y 14 casos de duplicaciones (22,5%) de uno o más exones. Se encontraron 5 duplicaciones de un solo nucleótido y 2 deleciones, de uno y 5 nucleótidos. La relación entre duplicación y deleción fue de 0,41 (15/34), siendo las deleciones 2 veces más frecuentes que las duplicaciones, evidenciando que la deleción y la duplicación no son eventos recíprocos.
Las deleciones de un solo exón se presentaron en 12 casos (19%), y las deleciones de más de un exón, en 21 sujetos (33%). Las grandes deleciones (más de 10 exones) se presentaron en 6 casos (9,6%). El 42% de la muestra presenta deleciones grandes entre los exones 45 y 54. De todos los exones, el 47 fue el más frecuentemente suprimido. Se observó un caso que presentó una gran deleción de 61 exones. El 50% de los casos presentó duplicaciones entre los exones 5 y 16. Las duplicaciones de un solo exón se presentaron en 5 casos (8%). Las duplicaciones de menos de 6 exones se representaron en 10 casos (16%). No se encontraron duplicaciones en los exones 67-79. Se encontró un caso de duplicación en tándem (duplicación de los exones 5-22 y 46-52) (figs. 1 y 2).
 representan las diversas isoformas del gen. Es evidente que el espectro de las deleciones se extiende a lo largo del gen, sin embargo, se aprecia una mayor frecuencia de estas alrededor de los exones 37 a 55. Las deleciones afectan también dichas isoformas (por ejemplo, isoforma Dp71 afectada por deleciones en exones 62-79) explicando, probablemente, el retardo del desarrollo psicomotor y el déficit cognitivo y que estas manifestaciones no solo se deban, de manera secundaria al déficit propio originado en la alteración muscular.")
Deleciones detectadas en los sujetos analizados.
La barra numerada representa el gen de la distrofina con cada uno de sus exones. Se aprecia la distribución de las deleciones detectadas en los pacientes. Las líneas superiores (encima del gen) representan las diversas isoformas del gen. Es evidente que el espectro de las deleciones se extiende a lo largo del gen, sin embargo, se aprecia una mayor frecuencia de estas alrededor de los exones 37 a 55. Las deleciones afectan también dichas isoformas (por ejemplo, isoforma Dp71 afectada por deleciones en exones 62-79) explicando, probablemente, el retardo del desarrollo psicomotor y el déficit cognitivo y que estas manifestaciones no solo se deban, de manera secundaria al déficit propio originado en la alteración muscular.
 representan las diversas isoformas del gen. Las mutaciones puntuales en negrita indican las mutaciones que no había sido descritas previamente. El espectro mutacional se extiende a lo largo de todo el gen. Como en el caso de las deleciones, las duplicaciones y las mutaciones puntuales afectan también dichas isoformas, contribuyendo al fenotipo variable que incluye el déficit psicomotor y el déficit cognitivo de algunos de los casos aquí reportados.")
Duplicaciones y mutaciones puntuales detectadas en los sujetos analizados.
La barra numerada representa el gen de la distrofina con cada uno de sus exones. Se aprecia la distribución de las duplicaciones y mutaciones puntuales detectadas. Las líneas superiores (encima del gen) representan las diversas isoformas del gen. Las mutaciones puntuales en negrita indican las mutaciones que no había sido descritas previamente. El espectro mutacional se extiende a lo largo de todo el gen. Como en el caso de las deleciones, las duplicaciones y las mutaciones puntuales afectan también dichas isoformas, contribuyendo al fenotipo variable que incluye el déficit psicomotor y el déficit cognitivo de algunos de los casos aquí reportados.
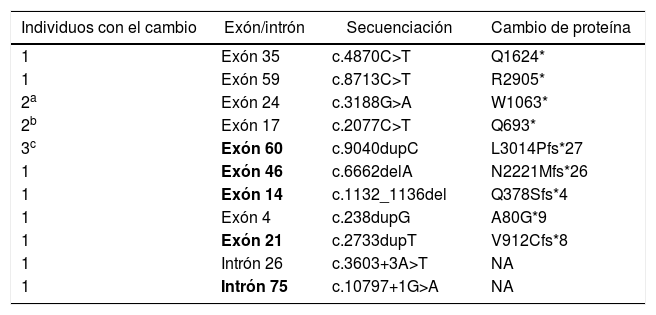
El estudio de secuenciación del gen DMD por la técnica de secuenciación masiva en paralelo fue realizado en 15 individuos (24,2%) con MLPA negativo. Se evidenciaron mutaciones sin sentido (nonsense) en 6 casos (9,6%), mutaciones que afectan el marco de lectura en 7 casos (11%) y mutaciones que afectan el splicing en 2 probandos (3,2%). No se encontraron mutaciones missense. Las variantes se distribuyeron a lo largo del gen y se encontraron 5 variantes que no habían sido previamente reportadas, identificadas en 7 individuos (tabla 2). La descripción de todas las mutaciones se aprecia en la tabla 3.
Resultados de secuenciación de 15 individuos con MLPA (n=63)
| Individuos con el cambio | Exón/intrón | Secuenciación | Cambio de proteína |
|---|---|---|---|
| 1 | Exón 35 | c.4870C>T | Q1624* |
| 1 | Exón 59 | c.8713C>T | R2905* |
| 2a | Exón 24 | c.3188G>A | W1063* |
| 2b | Exón 17 | c.2077C>T | Q693* |
| 3c | Exón 60 | c.9040dupC | L3014Pfs*27 |
| 1 | Exón 46 | c.6662delA | N2221Mfs*26 |
| 1 | Exón 14 | c.1132_1136del | Q378Sfs*4 |
| 1 | Exón 4 | c.238dupG | A80G*9 |
| 1 | Exón 21 | c.2733dupT | V912Cfs*8 |
| 1 | Intrón 26 | c.3603+3A>T | NA |
| 1 | Intrón 75 | c.10797+1G>A | NA |
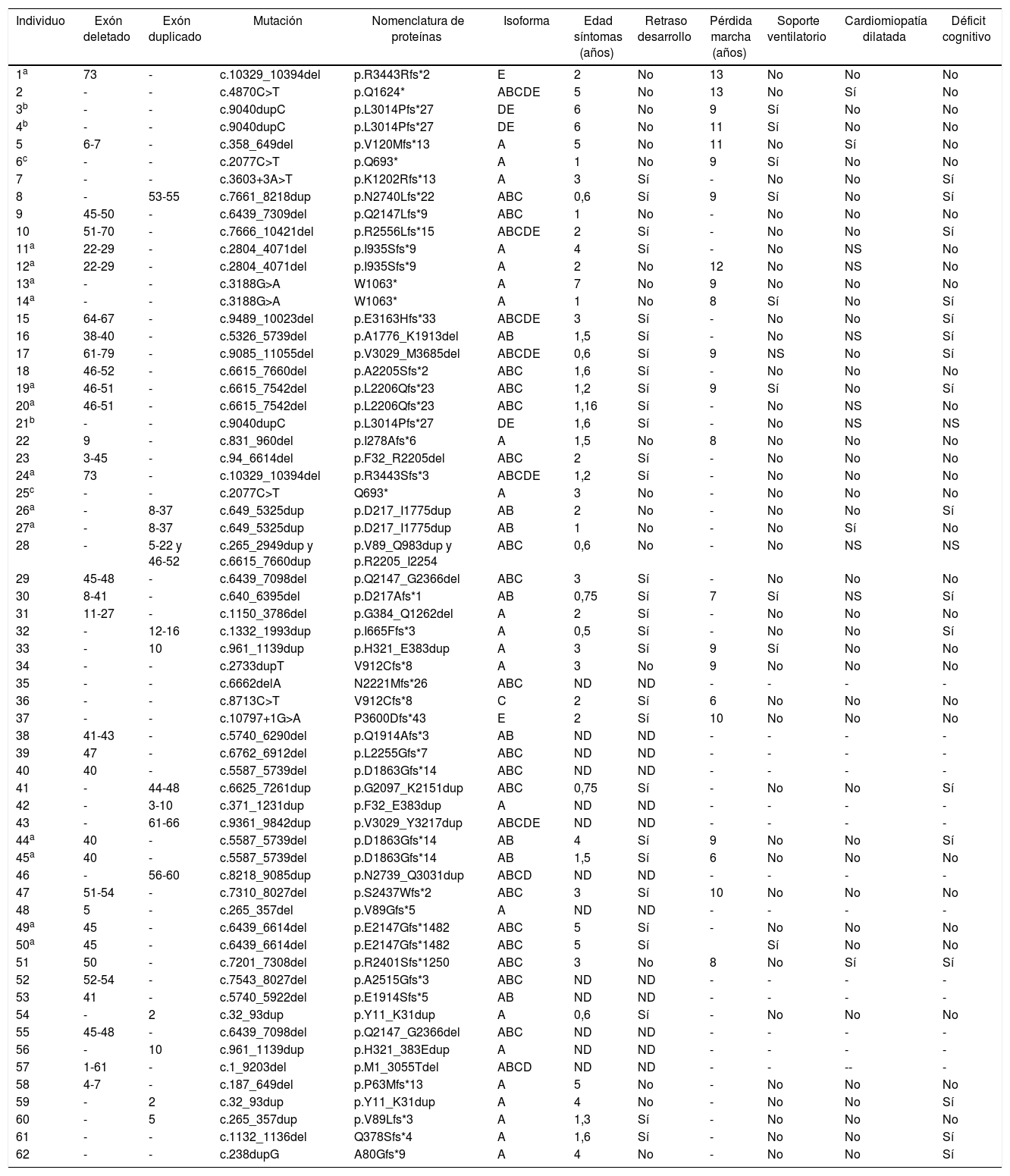
Descripción molecular y aspectos clínicos principales del grupo de pacientes
| Individuo | Exón deletado | Exón duplicado | Mutación | Nomenclatura de proteínas | Isoforma | Edad síntomas (años) | Retraso desarrollo | Pérdida marcha (años) | Soporte ventilatorio | Cardiomiopatía dilatada | Déficit cognitivo |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 73 | - | c.10329_10394del | p.R3443Rfs*2 | E | 2 | No | 13 | No | No | No |
| 2 | - | - | c.4870C>T | p.Q1624* | ABCDE | 5 | No | 13 | No | Sí | No |
| 3b | - | - | c.9040dupC | p.L3014Pfs*27 | DE | 6 | No | 9 | Sí | No | No |
| 4b | - | - | c.9040dupC | p.L3014Pfs*27 | DE | 6 | No | 11 | Sí | No | No |
| 5 | 6-7 | - | c.358_649del | p.V120Mfs*13 | A | 5 | No | 11 | No | Sí | No |
| 6c | - | - | c.2077C>T | p.Q693* | A | 1 | No | 9 | Sí | No | No |
| 7 | - | - | c.3603+3A>T | p.K1202Rfs*13 | A | 3 | Sí | - | No | No | Sí |
| 8 | - | 53-55 | c.7661_8218dup | p.N2740Lfs*22 | ABC | 0,6 | Sí | 9 | Sí | No | Sí |
| 9 | 45-50 | - | c.6439_7309del | p.Q2147Lfs*9 | ABC | 1 | No | - | No | No | No |
| 10 | 51-70 | - | c.7666_10421del | p.R2556Lfs*15 | ABCDE | 2 | Sí | - | No | No | Sí |
| 11a | 22-29 | - | c.2804_4071del | p.I935Sfs*9 | A | 4 | Sí | - | No | NS | No |
| 12a | 22-29 | - | c.2804_4071del | p.I935Sfs*9 | A | 2 | No | 12 | No | NS | No |
| 13a | - | - | c.3188G>A | W1063* | A | 7 | No | 9 | No | No | No |
| 14a | - | - | c.3188G>A | W1063* | A | 1 | No | 8 | Sí | No | Sí |
| 15 | 64-67 | - | c.9489_10023del | p.E3163Hfs*33 | ABCDE | 3 | Sí | - | No | No | Sí |
| 16 | 38-40 | - | c.5326_5739del | p.A1776_K1913del | AB | 1,5 | Sí | - | No | NS | Sí |
| 17 | 61-79 | - | c.9085_11055del | p.V3029_M3685del | ABCDE | 0,6 | Sí | 9 | NS | No | Sí |
| 18 | 46-52 | - | c.6615_7660del | p.A2205Sfs*2 | ABC | 1,6 | Sí | - | No | No | No |
| 19a | 46-51 | - | c.6615_7542del | p.L2206Qfs*23 | ABC | 1,2 | Sí | 9 | Sí | No | Sí |
| 20a | 46-51 | - | c.6615_7542del | p.L2206Qfs*23 | ABC | 1,16 | Sí | - | No | NS | No |
| 21b | - | - | c.9040dupC | p.L3014Pfs*27 | DE | 1,6 | Sí | - | No | NS | NS |
| 22 | 9 | - | c.831_960del | p.I278Afs*6 | A | 1,5 | No | 8 | No | No | No |
| 23 | 3-45 | - | c.94_6614del | p.F32_R2205del | ABC | 2 | Sí | - | No | No | No |
| 24a | 73 | - | c.10329_10394del | p.R3443Sfs*3 | ABCDE | 1,2 | Sí | - | No | No | No |
| 25c | - | - | c.2077C>T | Q693* | A | 3 | No | - | No | No | No |
| 26a | - | 8-37 | c.649_5325dup | p.D217_I1775dup | AB | 2 | No | - | No | No | Sí |
| 27a | - | 8-37 | c.649_5325dup | p.D217_I1775dup | AB | 1 | No | - | No | Sí | No |
| 28 | - | 5-22 y 46-52 | c.265_2949dup y c.6615_7660dup | p.V89_Q983dup y p.R2205_I2254 | ABC | 0,6 | No | - | No | NS | NS |
| 29 | 45-48 | - | c.6439_7098del | p.Q2147_G2366del | ABC | 3 | Sí | - | No | No | No |
| 30 | 8-41 | - | c.640_6395del | p.D217Afs*1 | AB | 0,75 | Sí | 7 | Sí | NS | Sí |
| 31 | 11-27 | - | c.1150_3786del | p.G384_Q1262del | A | 2 | Sí | - | No | No | No |
| 32 | - | 12-16 | c.1332_1993dup | p.I665Ffs*3 | A | 0,5 | Sí | - | No | No | Sí |
| 33 | - | 10 | c.961_1139dup | p.H321_E383dup | A | 3 | Sí | 9 | Sí | No | No |
| 34 | - | - | c.2733dupT | V912Cfs*8 | A | 3 | No | 9 | No | No | No |
| 35 | - | - | c.6662delA | N2221Mfs*26 | ABC | ND | ND | - | - | - | - |
| 36 | - | - | c.8713C>T | V912Cfs*8 | C | 2 | Sí | 6 | No | No | No |
| 37 | - | - | c.10797+1G>A | P3600Dfs*43 | E | 2 | Sí | 10 | No | No | No |
| 38 | 41-43 | - | c.5740_6290del | p.Q1914Afs*3 | AB | ND | ND | - | - | - | - |
| 39 | 47 | - | c.6762_6912del | p.L2255Gfs*7 | ABC | ND | ND | - | - | - | - |
| 40 | 40 | - | c.5587_5739del | p.D1863Gfs*14 | ABC | ND | ND | - | - | - | - |
| 41 | - | 44-48 | c.6625_7261dup | p.G2097_K2151dup | ABC | 0,75 | Sí | - | No | No | Sí |
| 42 | - | 3-10 | c.371_1231dup | p.F32_E383dup | A | ND | ND | - | - | - | - |
| 43 | - | 61-66 | c.9361_9842dup | p.V3029_Y3217dup | ABCDE | ND | ND | - | - | - | - |
| 44a | 40 | - | c.5587_5739del | p.D1863Gfs*14 | AB | 4 | Sí | 9 | No | No | Sí |
| 45a | 40 | - | c.5587_5739del | p.D1863Gfs*14 | AB | 1,5 | Sí | 6 | No | No | No |
| 46 | - | 56-60 | c.8218_9085dup | p.N2739_Q3031dup | ABCD | ND | ND | - | - | - | - |
| 47 | 51-54 | - | c.7310_8027del | p.S2437Wfs*2 | ABC | 3 | Sí | 10 | No | No | No |
| 48 | 5 | - | c.265_357del | p.V89Gfs*5 | A | ND | ND | - | - | - | - |
| 49a | 45 | - | c.6439_6614del | p.E2147Gfs*1482 | ABC | 5 | Sí | - | No | No | No |
| 50a | 45 | - | c.6439_6614del | p.E2147Gfs*1482 | ABC | 5 | Sí | Sí | No | No | |
| 51 | 50 | - | c.7201_7308del | p.R2401Sfs*1250 | ABC | 3 | No | 8 | No | Sí | Sí |
| 52 | 52-54 | - | c.7543_8027del | p.A2515Gfs*3 | ABC | ND | ND | - | - | - | - |
| 53 | 41 | - | c.5740_5922del | p.E1914Sfs*5 | AB | ND | ND | - | - | - | - |
| 54 | - | 2 | c.32_93dup | p.Y11_K31dup | A | 0,6 | Sí | - | No | No | No |
| 55 | 45-48 | - | c.6439_7098del | p.Q2147_G2366del | ABC | ND | ND | - | - | - | - |
| 56 | - | 10 | c.961_1139dup | p.H321_383Edup | A | ND | ND | - | - | - | - |
| 57 | 1-61 | - | c.1_9203del | p.M1_3055Tdel | ABCD | ND | ND | - | - | -- | - |
| 58 | 4-7 | - | c.187_649del | p.P63Mfs*13 | A | 5 | No | - | No | No | No |
| 59 | - | 2 | c.32_93dup | p.Y11_K31dup | A | 4 | No | - | No | No | Sí |
| 60 | - | 5 | c.265_357dup | p.V89Lfs*3 | A | 1,3 | Sí | - | No | No | No |
| 61 | - | - | c.1132_1136del | Q378Sfs*4 | A | 1,6 | Sí | - | No | No | Sí |
| 62 | - | - | c.238dupG | A80Gfs*9 | A | 4 | No | - | No | No | Sí |
Isoformas de la distrofina: A=Dp427; B=Dp260; C=Dp140; D=Dp116; E=Dp71.
La DMD es una miopatía de origen genético de herencia recesiva ligada a X, causada por mutaciones en el gen de la distrofina (Xp21.2). Afecta principalmente a los hombres, sin embargo, las mujeres portadoras pueden manifestar debilidad muscular y elevación de niveles de CK30. Los pacientes incluidos en el presente análisis fueron exclusivamente hombres. En este estudio, las mutaciones patogénicas en el gen de la distrofina se identificaron en 62 individuos colombianos con sospecha clínica de DMD/DMB: un 53,3% de los casos por deleción, un 22,5% de los casos por duplicación y un 24,2% de los casos con mutación puntual. En 7 de los probandos (11%) se identificaron mutaciones puntuales que no habían sido previamente reportadas.
Un estudio previo realizado en Colombia detectó un porcentaje menor de deleciones (31%), sin embargo, dicho estudio utilizó únicamente PCR múltiple (17 exones y promotor)31. Un estudio reciente del espectro mutacional de la DMD en España reportó deleciones en el 46% de los casos32, mientras que otro estudio en la India reportó deleciones en el 68% de los casos utilizando MLPA33, similar a lo reportado en Polonia (65%)34, mientras que en Brasil una cohorte presentó un 58% de deleciones35. El menor porcentaje de la serie española puede deberse a un sesgo de selección según los autores32. A pesar de la variación de serie a serie, las deleciones son la principal causa de mutación del gen de la DMD/DMB. En general, se considera que las grandes deleciones del gen son responsables de más del 65% de los casos de DMD36. Las deleciones no tienen una distribución al azar a lo largo del gen, sino que se agrupan en regiones proclives o hot spots. Alrededor del 80% de las deleciones se concentran entre los exones 44 y 5237 y alrededor del 20% comprende los exones 1 al 1938, similar a lo descrito en el presente estudio. Las duplicaciones contribuyen con alrededor del 14% de los casos39 y entre el 10 y el 30% se deben a pequeñas deleciones o inserciones, mutaciones puntuales o defectos en el splicing40. En la base global de mutaciones DMD (TREAT-NMD DMD)39 se ha identificado que alrededor del 10% de las causas de la enfermedad se deben a mutaciones puntuales; la descripción de mutaciones nuevas como las aquí descritas contribuye a dibujar el espectro mutacional de la enfermedad.
El fenotipo depende de la localización de la mutación en el gen y no necesariamente del tamaño de la deleción o de la duplicación; una deleción que afecte el marco de lectura, independiente del tamaño, produce una mutación severa, mientras que una deleción que no afecte el marco de lectura produce un fenotipo más leve asociado con DMB4. Entre los primeros signos para sospechar DMD se incluyen el retraso en la marcha, la marcha inestable y en puntas, la dificultad para subir escaleras, la debilidad muscular progresiva, la pseudohipertrofia de gastrocnemios y la incapacidad para levantarse desde el suelo sin usar los brazos (signo de Gowers)41. En la mayoría de los probandos de este estudio se ha descrito el antecedente de retraso en el desarrollo psicomotor, lo que se puede explicar tanto por el componente de déficit muscular como por el posible papel de la distrofina en el desarrollo y la perfusión cerebral y como agente neuroinmunomodulador42. Las mutaciones en el gen DMD pueden conducir a un largo espectro de fenotipos clínicos que están estrechamente correlacionados con las alteraciones moleculares y con la severidad de la alteración en la función proteica causada por la mutación43, sin embargo, la correlación fenotipo-genotipo no es directa, en especial los aspectos relacionados con las manifestaciones neurológicas, debido principalmente a las 6 isoformas cerebrales de la distrofina, las cuales juegan un papel relevante en la maduración y el remodelamiento de las sinapsis neuronales44. En el presente estudio se logró determinar que en 28 casos (45%) el retraso del desarrollo estaba presente como antecedente, sin embargo, este hallazgo puede ser altamente inespecífico, debido a la diversidad de manejos recibidos (terapias físicas) o manifestaciones sutiles no identificadas previamente. Los signos clínicos descritos en el grupo estudiado forman parte del espectro clínico usualmente reportado, sin embargo, no es posible hacer una adecuada correlación fenotipo-genotipo debido a que fueron remitidos con diversos tiempos de evolución del cuadro clínico y con tratamientos disímiles.
La correlación fenotipo-genotipo es incierta; a pesar de que se conoce la localización de las isoformas y en algunos casos su función, la deleción de ciertas regiones no se comporta de forma similar en todos los pacientes. Se ha descrito que los pacientes con compromiso de regiones terminales del gen DMD (exones 62-79) por alteración de la isoforma Dp71 cursan con déficit cognitivo, esto debido al potencial de afectar la expresión de un número creciente de isoformas por efectos perjudiciales acumulativos45; en nuestra cohorte el 50% de los individuos con deleción de la parte distal de la proteína (6 pacientes) cursaban con déficit cognitivo. Respecto al resto de las isoformas comprometidas y el fenotipo, no se encontró una relación específica fenotipo-genotipo.
Aunque la biopsia muscular (inmunohistoquímica o inmunoblot) juega un papel importante en el diagnóstico46, la caracterización molecular de la enfermedad es necesaria para la consejería genética, el pronóstico y para el eventual manejo farmacológico de ciertas mutaciones. El diagnóstico molecular de la DMD incluye la PCR múltiple, sin embargo, esta solo detecta deleciones en un grupo de exones, no detecta rearreglos genéticos ni mutaciones puntuales. Algunas de estas falencias se superan con la técnica de MLPA, que identifica deleciones/duplicaciones en los 79 exones del gen DMD, delimita el tamaño de estas mutaciones y determina el estado de portador en las mujeres. Diversos estudios han mostrado su beneficio diagnóstico frente a la PCR múltiple47.
En el caso de pacientes con sospecha clínica de DMD, sin evidencia de deleción/duplicación, se debe optar por la secuenciación del gen, en donde se evalúan variantes en la secuencia exónica; sin embargo, no detecta mutaciones en el 2% de los casos con rearreglos complejos o cambios intrónicos profundos48. Actualmente solo se dispone de manejo paliativo para la DMD, el cual se basa en terapia física, el uso de apoyos ortopédicos, el tratamiento de las complicaciones y la administración de glucocorticoides. Algunos estudios clínicos han indicado que los glucocorticoides pueden prolongar la marcha y mejorar la función y la fortaleza muscular13,14. Sin embargo, generan varios efectos adversos, sin modificar la función de la proteína distrofina y, por ende, sin impactar significativamente sobre la etiología de la enfermedad14.
En este contexto, han surgido terapias potenciales para la DMD, siendo las más prometedoras los oligonucleótidos antisentido que inducen salto exonal15–17 y las terapias de salto de codón de parada temprano originadas en mutaciones sin sentido18,49. Es así como es necesaria la caracterización molecular de la enfermedad, tanto para la consejería genética como para evaluar el uso de manejo farmacológico19. Por otra parte, la caracterización por grupos poblacionales permite establecer prevalencias globales y comparar las frecuencias de variantes genéticas entre grupos humanos20.
El diagnóstico molecular de mutaciones puntuales a través de la secuenciación masiva paralela permitió alcanzar de manera eficiente la confirmación diagnóstica, definiendo para los casos negativos para deleción/duplicación la asesoría adecuada y la implementación de un manejo terapéutico apropiado.
AutoríaMGA: diseño del estudio, recogida y análisis de los datos, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
TP: diseño del estudio, recogida y análisis de los datos, realización de pruebas moleculares, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
MGT: diseño del estudio, recogida y análisis de los datos, redacción del artículo, valoración clínica de los sujetos y realización de árboles familiares, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
RGR: diseño del estudio, recogida y análisis de los datos, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
PAR: diseño del estudio, recogida y análisis de los datos, realización de pruebas moleculares y manejo de muestras, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
TB: diseño del estudio, recogida y análisis de los datos, realización de pruebas moleculares, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito, incluyendo la veracidad y la integridad de los datos y los resultados.
AP: diseño del estudio, recogida y análisis de los datos, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito.
FSO: diseño del estudio, recogida y análisis de los datos, redacción del artículo, aprobación de la versión final del mismo. Es responsable de todos los aspectos del manuscrito.
FinanciaciónEl presente estudio fue financiado en parte por una donación de los Laboratorios Vitalchem de Colombia y PTC Therapeutics y por el Instituto de Genética Humana de la Facultad de Medicina de la Pontificia Universidad Javeriana, Proyecto de investigación-Vicerrectoría de Investigación: 7034.
Conflicto de interesesNinguno que declarar por parte de los autores.
