





El trastorno por deficiencia de CDKL5 es una rara enfermedad en la que hay un inicio muy precoz de las convulsiones. Esta se relaciona con epilepsia refractaria, lo que complica su manejo terapéutico y puede empeorar el pronóstico de nuestros pacientes.
Caso clínicoMujer de 2años diagnosticada de trastorno por deficiencia de CDKL5 que presentó durante su estancia en nuestro centro, donde ingresó por mal control de su epilepsia, un estatus epiléptico no convulsivo refractario cuyo manejó se realizó con diversos anticomiciales, yugulando finalmente las crisis y la actividad eléctrica en el electroencefalograma tras la introducción de ketamina.
ConclusionesEl estatus epiléptico refractario constituye una urgencia neurológica que puede dejar graves secuelas en nuestros pacientes, por lo que debemos abordarlo de una manera adecuada y precoz. La ketamina podría ser un fármaco útil en el manejo, aunque se requieren más estudios sobre su uso en niños.
CDKL5 deficiency disorder is a rare disease in which there is a very early onset of seizures. This is related to refractory epilepsy, which complicates its therapeutic management and can worsen the prognosis of our patients.
Clinical caseA 2-year-old woman diagnosed with CDKL5 deficiency disorder, who presented during her stay in our center, where she was admitted due to poor control of her epilepsy, a refractory non-convulsive epileptic status, which was managed with various anticomial drugs, juggling the crises and the electrical activity in the electroencephalogram finally after the introduction of ketamine.
ConclusionsRefractory epileptic status constitutes a neurological emergency that can leave serious sequelae in our patients, so we must address it in an appropriate and early way. Ketamine could be a useful drug in management, although more studies on its use in children are required.
El síndrome de Rett (RTT) es una rara enfermedad, con una incidencia aproximada de 1/10.000 mujeres, que afecta al neurodesarrollo. Se caracteriza por un desarrollo normal durante los primeros 6-18meses de vida, seguidos de un período breve de estancamiento y posterior regresión de las capacidades cognitivas y psicomotoras, con pérdida de las habilidades del lenguaje y el uso de las manos, marcha alterada o ausente, dispraxia, déficits cognitivos y comportamientos estereotipados1,2.
Según los nuevos criterios revisados, se ha reconocido que algunas personas que presentan muchas de las características clínicas del RTT no se ajustan a los criterios establecidos para el diagnóstico del fenotipo clásico. Entre estos casos, conocidos inicialmente como RTT «atípicos», se encuentra la «variante de Hanefeld», actualmente denominada trastorno por deficiencia de CDKL5 (CDD). El CDD, cuya incidencia es inferior al RTT (1/40.000-1/60.000), se caracteriza por un retraso grave en el desarrollo desde el nacimiento y el inicio de las convulsiones antes de los 3meses, con alta prevalencia de trastornos del sueño, características que lo diferencian del RTT1,3.
Las mutaciones en el gen MeCP2 (Methyl CpG Binding Protein 2) ligado al X representan el 90-95% de los casos del RTT (causantes del fenotipo clásico), mientras que las mutaciones en el gen ligado aX que codifica la ciclina dependiente de quinasa5 (CDKL5), que actúa como regulador de MeCP2, con importantes implicaciones en el desarrollo cerebral y la maduración neuronal, causan CDD. En ambos casos se produce un deterioro en las respuestas sinápticas glutamatérgicas corticales y la conectividad excitadora, así como una disfunción del sistema gabaérgico4,5.
Caso clínicoMujer preescolar de 2años que acude a urgencias por episodios de crisis epilépticas generalizadas consistentes en crisis de hipertonía con movimientos tónico-clónicos de extremidades y mirada perdida, de minutos de duración, en número de 10 al día en las últimas 72h. Previamente la paciente presentaba 2-3 episodios al día.
La paciente presenta un retraso psicomotor severo con marcada hipotonía y conducta autística.
Como antecedentes personales, nuestra paciente fue diagnosticada de un CDD. Presentó una primera convulsión a los 3meses de edad, realizándose TC y RNM, que resultaron normales, así como EEG, en el que se evidenció una actividad paroxística focal en hemisferio derecho, por lo que se inició tratamiento con levetiracetam y ácido valproico, que se han mantenido hasta la actualidad, hasta alcanzar dosis de 50mg/kg/día de ambos fármacos. Resto de estudios realizados a los 16meses, incluido estudio metabólico, estudio genético de síndrome de Rett y esclerosis tuberosa negativos. En el estudio de exoma clínico se encuentra la variante patogénica c.543delp (Glu181Aspfs*47) en heterocigosis, del gen CDKL5 (autosómico dominante), no descrita previamente.
Durante el ingreso la paciente tuvo crisis recurrentes de escasos minutos de duración, con recuperación intercrítica completa, con las características ya descritas, que responden inicialmente a la administración de benzodiacepinas (midazolam y clonazepam). Analítica sanguínea con gasometría venosa sin alteraciones. Niveles de fármacos antiepilépticos en rango.
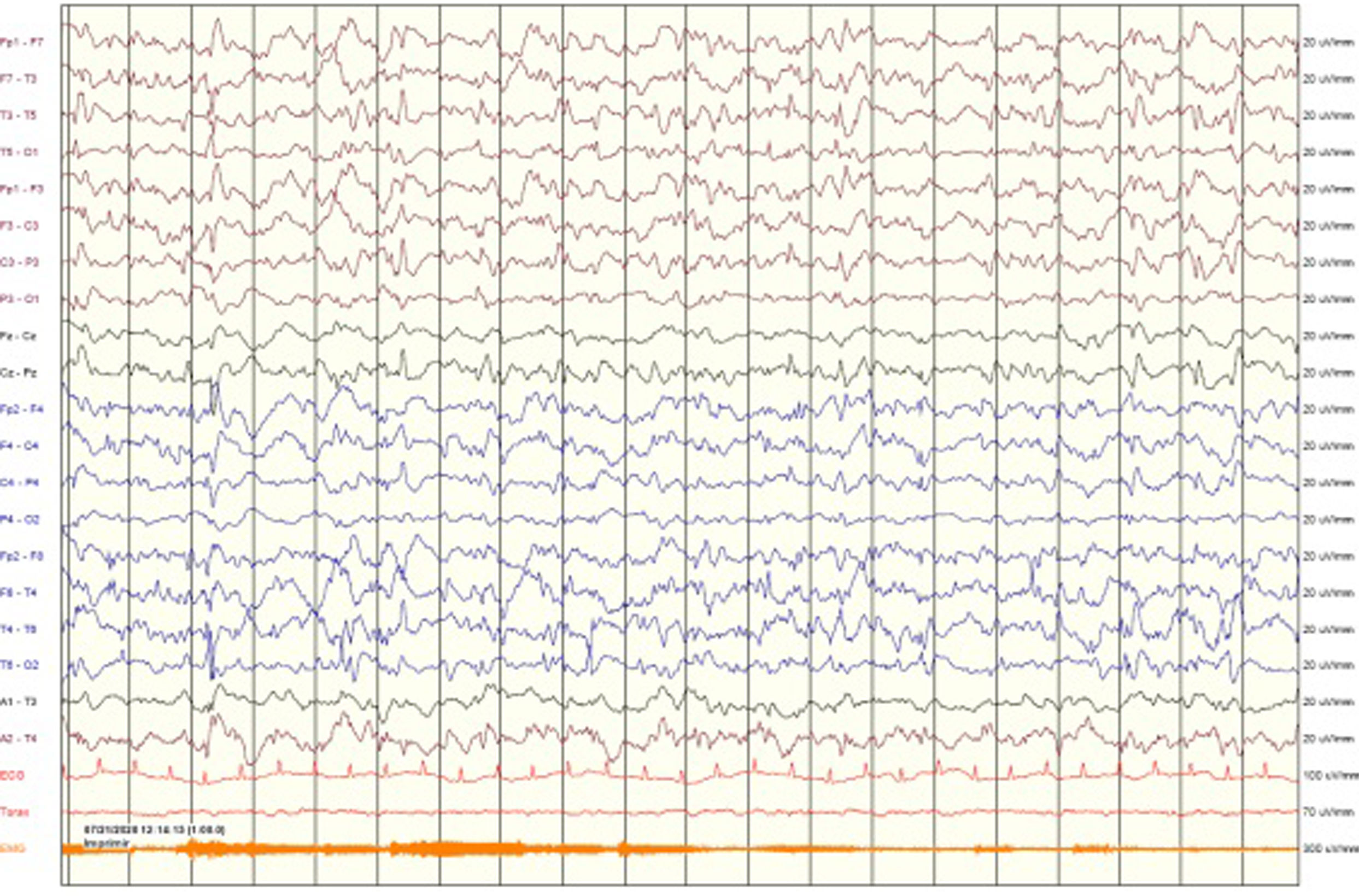
A las 18h de ingreso la paciente muestra un empeoramiento clínico con hipoactividad y crisis tónico-clónicas breves, recurrentes, sin recuperación entre ellas, quedando posteriormente arreactiva sin actividad convulsiva. Se realiza EEG, en el que se registra una actividad paroxística generalizada continua, compatible con estatus epiléptico (EE) no convulsivo (fig. 1). Se pauta nuevo bolo de midazolam i.v. a 0,2mg/kg y lancosamida a 8mg/kg, realizándose EEG de control en el que se observa persistencia del EE no convulsivo.

Se decide traslado a la unidad de cuidados intensivos pediátrica (UCIp) de hospital de referencia, sin precisar soporte respiratorio, para monitorización continua con EEG y tratamiento de EE refractario.
A su llegada a UCIp se inicia monitorización continua con EEG y perfusión de midazolam a 0,2mg/kg/h. Posteriormente se administran 2bolos de ketamina (KET) 10mg por convulsión clínica con correlato eléctrico, con buena respuesta en el EEG tras el segundo bolo (fig. 2).

La paciente pasa a planta tras cesar el EE, sin presentar nuevos episodios convulsivos, presentando recuperación progresiva del nivel de consciencia hasta normalización con actividad basal habitual. Es dada de alta al quinto día de ingreso, con levetiracetam y ácido valproico según pauta habitual. Ha realizado seguimiento en consultas de Neuropediatría, realizando mantenimiento con levetiracetam y ácido valproico, e inicio de perampanel.
DiscusiónLa epilepsia asociada al RTT es frecuentemente resistente al tratamiento y representa un problema clínico significativo en estos pacientes. En un gran estudio de pacientes con RTT la epilepsia estuvo presente en dos tercios (64,2%) de los pacientes con RTT de todo tipo, con epilepsia resistente al tratamiento en menos de un quinto (17,2%). La epilepsia está presente en todos aquellos con CDD3.
El EE, incluido el EE no convulsivo, se encuentra entre las emergencias neurológicas más comunes. El EE refractario se define como el EE que no responde a los fármacos antiepilépticos de primera y segunda línea utilizados adecuadamente6.
En cuanto a su fisiopatología, debemos destacar que, debido a cambios maladaptativos subcelulares, los fármacos acidérgicos γ-aminobutíricos (entre los que se encuentran las benzodiacepinas o el ácido valproico) ya no son efectivos, y hay un aumento en los receptores sinápticos de N-metil-d-aspartato (NMDA). La KET, fármaco ampliamente conocido como anestésico, es un antagonista no competitivo del receptor de NMDA que podría ser útil en el tratamiento del EE refractario de cualquier causa en niños y adultos, con una tasa de éxito del 73 y del 74%, respectivamente, según las series estudiadas, aunque existe poca experiencia publicada en el tratamiento de EE refractario con KET. Además, no se describieron efectos adversos significativos7.
Los bloqueadores de NMDA como la KET han sido efectivos en la supresión de EE incluso después del fracaso de las benzodiacepinas en experimentos con animales, según algunos estudios8.
En cuanto al uso de lacosamida, nuevo antiepiléptico de tercera generación que actúa facilitando la inactivación lenta de los canales de sodio, esta se muestra útil y segura en epilepsias focales refractarias al tratamiento y como terapia complementaria en pacientes mayores de 16años; su utilidad en el manejo del EE refractario pediátrico se postula como una buena opción como tratamiento adicional dada su eficacia en el control de las convulsiones y el perfil de seguridad, y a pesar de no resultar efectiva en nuestra paciente, podría considerarse como una opción válida, previo al uso de KET9.
ConclusionesEl CDD es una rara enfermedad que se caracteriza por epilepsia refractaria. Puede debutar muy precozmente y evolucionar a un EE, en ocasiones refractario, debido a su difícil manejo terapéutico, como en el caso de nuestra paciente, requiriendo incluso fármacos anestésicos de tercera línea para su manejo.
El EE no convulsivo refractario representa una emergencia neurológica compleja asociada a disfunción neurológica a largo plazo y alta mortalidad, por lo que un manejo terapéutico adecuado y precoz resulta fundamental. Los desafíos en el manejo surgen ya que la etiología subyacente no siempre se reconoce rápidamente y las opciones terapéuticas se limitan con las convulsiones prolongadas. La KET se postula como una opción terapéutica útil y segura en el manejo de estos pacientes, como exponemos en nuestro caso.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
