




La encefalitis por anticuerpos anti-receptor de NMDA afecta principalmente a personas jóvenes y suele requerir una alta sospecha clínica para su diagnóstico. Su forma de presentación típica se caracteriza por síntomas psiquiátricos, alteraciones del movimiento, crisis epilépticas y disautonomía. Presentamos un caso sumamente inusual, con afectación radiológica exclusiva del romboencéfalo y un síndrome pancerebeloso como expresión clínica.
Anti-NMDA receptor encephalitis mainly affects young people and it requires a high clinical suspicion for diagnosis. The typical clinical presentation included psychiatric symptoms, movement disorders, seizures and dysautonomia. We present a highly inusual case where the rhombencephalon was exclusively affected, leading to a clinical expression of a pancerebellar syndrome.
La encefalitis autoinmune mediada por anticuerpos anti-receptor de NMDA (anti-NMDAr) fue descubierta en el año 2007 por Dalmau et al.1. Se define como aquella producida por inmunoglobulinas IgG dirigidas contra la subunidad GluN1 del receptor N-metil-D-aspartato (NMDA)2. Su incidencia en la población general es desconocida, si bien algunos estudios la estiman en 1-5 casos por millón de habitantes al año3. Existe una predominancia clara en las mujeres (80% de los casos) y la edad media de presentación se sitúa en los 21 años3,4.
Determinados factores pueden actuar como desencadenantes de este tipo de encefalitis. Algunos ejemplos bien conocidos son determinados tipos de tumores y la encefalitis herpética3,4. La asociación con neoplasias dependerá de la edad y el sexo, siendo el teratoma de ovario el más frecuente, principalmente en mujeres de más de 18 años2,4. En el caso de los varones o los mayores de 45 años de edad, el hallazgo de tumores es infrecuente, si bien se han descrito casos asociados a tumores de células germinales, teratomas mediastínicos, carcinomas de pulmón de células pequeñas, linfoma de Hodgkin y neuroblastoma4.
En muchos pacientes, la presentación clínica típica viene precedida unos días antes por un síndrome prodrómico que puede simular una infección vírica3,4. Más adelante, con frecuencia se desarrollan síntomas psiquiátricos (psicosis, manía, depresión, catatonia…), alteraciones del movimiento, crisis epilépticas o disautonomía3,4. Las manifestaciones clínicas ocasionadas por la afectación del romboencéfalo, como las que se describen en este artículo, son mucho menos frecuentes2,5,6.
El diagnóstico de sospecha se basa en la clínica apoyada en los resultados de las pruebas complementarias. El LCR suele mostrar pleocitosis de predominio mononuclear (80%), hiperproteinorraquia (30%) o bandas oligoclonales (60%). Los estudios electroencefalográficos pueden detectar un registro enlentecido y desorganizado o actividad epiléptica. La RM cerebral suele ser poco expresiva, pues solo en el 35% de las ocasiones se podrán observar alteraciones de la señal. Para el diagnóstico de confirmación, no obstante, es necesaria la detección en LCR de anticuerpos IgG contra la subunidad GluN1 del receptor NMDA3,4.
El tratamiento de primera línea recomendado son esteroides, inmunoglobulinas inespecíficas o plasmaféresis, acompañados cuando la situación lo requiera de la extirpación del tumor3,4.
En general, el pronóstico es bueno si se administra el tratamiento adecuado de forma precoz. No obstante, un 10-25% de los casos sufren recidivas en los primeros años3,4.
Caso clínicoUn varón de 21 años de edad, sin antecedentes personales de interés, fue remitido al hospital por un cuadro clínico de curso progresivo y aproximadamente una semana de evolución consistente en mareos, cefalea opresiva de localización occipital y diplopía binocular. Durante el mes anterior había acudido en diversas ocasiones a su médico de Atención Primaria por molestias abdominales, náuseas y vómitos, por lo que se le había prescrito tratamiento con omeprazol, metoclopramida y betahistina, lográndose un alivio parcial de los síntomas.
En la exploración física el día de su ingreso presentaba un estado de letargia, discreta limitación de la abducción del ojo derecho, ataxia apendicular bilateral asimétrica de predominio izquierdo e hiporreflexia generalizada.
Los análisis de sangre, las radiografías de tórax y la TC cerebral sin contraste no mostraron hallazgos significativos. Se efectuó una punción lumbar que dio salida a un LCR de aspecto claro y presión de apertura normal. El estudio citoquímico reveló una pleocitosis de predominio mononuclear (53 linfocitos/mm3) e hiperproteinorraquia (1,01g/l), sin consumo de glucosa. Las pruebas microbiológicas (tinción de Gram y cultivo) y las PCR de las principales bacterias, hongos y virus neurotropos (Mycobacterium tuberculosis, Listeria monocytogenes, Mycoplasma pneumoniae, enterovirus, citomegalovirus, virus de Epstein-Barr, virus herpes simple tipo 1 y 2, virus herpes 6, virus varicela zóster, Cryptococcus neoformans, entre otros) fueron negativas.
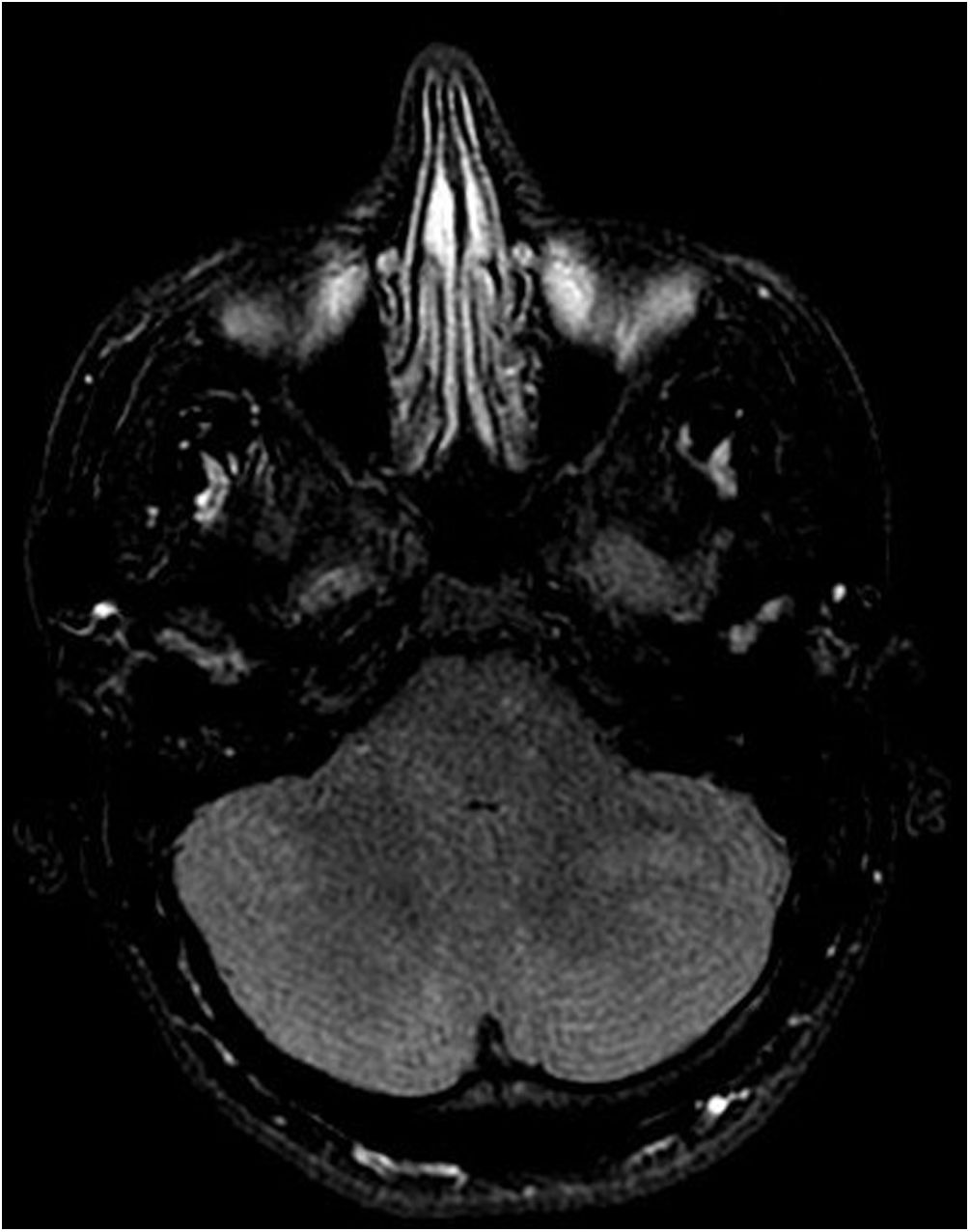
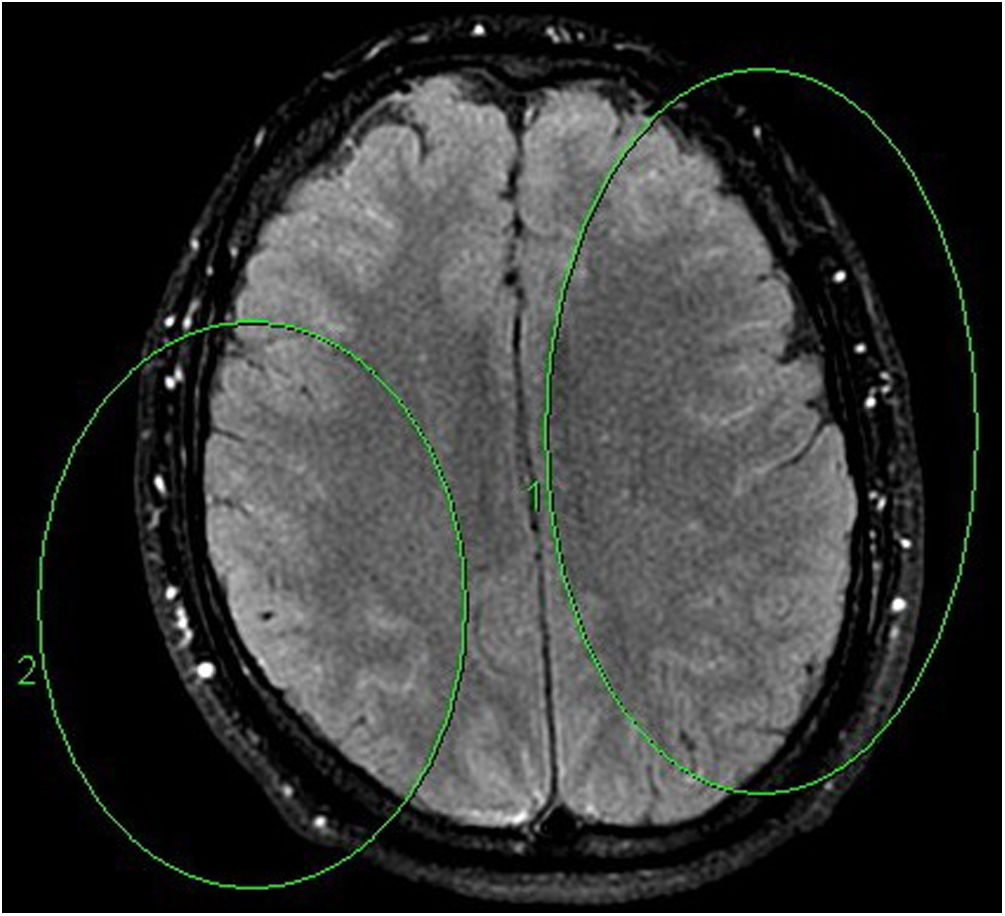
En la RM cerebral con contraste se observaron áreas de aumento de la señal en secuencias T2/Flair localizadas en los hemisferios cerebelosos y la protuberancia (fig. 1), así como una hipercaptación meníngea (fig. 2). Los estudios neurofisiológicos de conducción nerviosa fueron informados como normales.


Ante la sospecha clínica de meningoencefalitis se administró tratamiento con aciclovir y ampicilina endovenosos y, posteriormente, con inmunoglobulinas inespecíficas (0,4g/kg de peso/día) durante 5 días, sin que se observara ninguna mejoría clínica.
El tercer día de ingreso sufrió un episodio de agitación psicomotriz, seguido de una crisis convulsiva tónico-clónica y un periodo prolongado de disminución del nivel de consciencia. Se trasladó a la Unidad de Cuidados Intensivos, donde permaneció 11 días sedado e intubado. En este tiempo se realizaron electroencefalogramas seriados, que mostraron un enlentecimiento de la actividad cerebral de base con brotes de ondas delta de gran amplitud en las regiones frontales. El día 14 se retiró la medicación sedante y se observó un empeoramiento clínico en forma de un síndrome pancerebeloso compuesto por dismetría ocular, habla escandida, ataxia apendicular bilateral simétrica y ataxia troncular.
Se administraron bolos de 6-metilprednisolona (1g diario) durante 3 días sin apreciarse respuesta clínica. Se prorrogó esta terapia 5 días más y por fin se evidenció cierta mejoría del síndrome. Secuencialmente se pautó un nuevo ciclo de inmunoglobulinas inespecíficas (0,4g/kg de peso/día) durante 5 días y se consolidaría la evolución favorable. Un mes después del alta hospitalaria, el paciente podía llevar una vida autónoma, encontrándose paucisintomático.
Entre los estudios diagnósticos se incluyeron diversas pruebas dirigidas a excluir un síndrome paraneoplásico (TC de cuello, tórax, abdomen y pelvis, ecografía testicular, PET-TC con 18-FDG), entre cuyos resultados solamente destacaba un granuloma pulmonar aislado y esplenomegalia. Un análisis de líquido broncoalveolar no fue compatible con una sarcoidosis. Por otra parte, los análisis de sangre y LCR incluyeron múltiples serologías y estudios de autoinmunidad de enfermedades sistémicas y específicas del sistema nervioso, sin que se lograsen detectar hallazgos que pudieran relacionarse con la enfermedad.
Un análisis de anticuerpos en un laboratorio externo demostró la presencia en el LCR de anticuerpos dirigidos contra el receptor NMDA, así como una reactividad adicional anómala que fue interpretada como un segundo anticuerpo contra antígenos de superficie, cuya identidad no ha podido ser caracterizada hasta la fecha.
Discusión y conclusionesEl objetivo de esta presentación es aumentar la casuística y ampliar el espectro clínico de esta enfermedad. Se trata de un caso de encefalitis autoinmune mediada por anticuerpos anti-NMDAr de presentación atípica. En la literatura científica publicada hasta la fecha se han descrito varios casos en los que la afectación del tronco del encéfalo o el cerebelo forman parte del síndrome ocasionado por los anticuerpos anti-NMDAr2, pero la presencia de una romboencefalitis aislada como presentación inicial de la enfermedad está escasamente documentada5,6.
La presencia en el LCR de anticuerpos IgG contra la subunidad GluN1 del receptor NMDA se considera diagnóstica de esta patología, siendo su especificidad cercana al 100%. En cambio, un 10% de los pacientes no poseen este tipo de anticuerpos en el suero y existen casos diagnosticados erróneamente de encefalitis anti-NMDAr por la existencia de falsos positivos séricos3,4. Por otra parte, desconocemos la implicación que podría tener en la patología del paciente el segundo anticuerpo contra antígenos de superficie detectado en las muestras de LCR. Algunos estudios afirman que un 4-7,5% de las encefalitis anti-NMDAr podrían presentar anticuerpos coexistentes, siendo esta un área de investigación en expansión7.
En el futuro, la notificación de casos con características comparables podría ayudar a conocer la incidencia de la romboencefalitis en la enfermedad mediada por anticuerpos anti-NMDAr y la importancia que podrían tener en su patogenia otros anticuerpos coexistentes.
FinanciaciónLa presente publicación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no presentar ningún conflicto de interés.
