





El síndrome de Miller Fisher se caracteriza principalmente por la tríada de ataxia, oftalmoplejía e hiporreflexia.
Caso clínicoPresentamos el caso de un paciente que experimentó estos síntomas de forma atípica y progresiva. Se realizó pesquisa imagenológica y paraclínica para descartar los diagnósticos diferenciales con mayores probabilidades epidemiológicas y realizar el diagnóstico de SMF. El tratamiento consistió en plasmaféresis, logrando mejoría clínica.
ConclusionesEl objetivo de este reporte es compartir nuestra experiencia en el enfoque diagnóstico del SMF, lo cual es esencial para iniciar un tratamiento óptimo de manera temprana y mejorar el pronóstico de los pacientes.
Miller Fisher syndrome (MFS) is primarily characterized by the triad of ataxia, ophthalmoplegia, and hyporeflexia.
Clinical caseWe present the case of a patient who experienced these symptoms in an atypical and progressive manner. Imaging and paraclinical investigations were conducted to rule out differential diagnoses with higher epidemiological probabilities and establish the diagnosis of MFS. The treatment consisted of plasma exchange, resulting in clinical improvement.
ConclusionsThe objective of this report is to share our experience in the diagnostic approach to MFS, which is essential for initiating optimal early treatment and improving the prognosis of patients.
El síndrome de Miller Fisher (SMF) representa la variante clínica más frecuente del síndrome de Guillain-Barré (SGB) con un 10-20% del total de casos en países occidentales y hasta a un 25% en población japonesa1. Se trata de una polineuropatía monofásica que se presenta como un cuadro agudo y autolimitado consistente en una tríada típica de síntomas: oftalmoplejía, ataxia y arreflexia, usualmente precedidos de síntomas respiratorios altos o gastrointestinales2-3; sin embargo, puede tener presentaciones limitadas como una oftalmoplejía bilateral o unilateral sin otros síntomas asociados4. De igual manera, se han reportado otros signos y síntomas además de la presentación clásica, tales como neuropatía óptica, parálisis facial tardía, cefalea, disgeusia y dificultad miccional5.
La etiopatogenia del SMF se basa en una respuesta autoinmune anómala posterior a una infección microbiana, generalmente producida por Campylobacter jejuni. Se considera que una reacción cruzada entre los antígenos de los nervios periféricos y epítopos bacterianos a través del mimetismo molecular, desencadena la producción de anticuerpos antigangliósidos GQ1b, los cuales contribuyen en el proceso inflamatorio de este trastorno4-6. Debido a su naturaleza autolimitada, esta variante tiende a tener una buena evolución clínica, con una resolución completa de los síntomas al cabo de aproximadamente seis meses. Su tratamiento consiste en sesiones de plasmaféresis o administración intravenosa de inmunoglobulina, con el objetivo de acelerar la recuperación y disminuir la probabilidad de complicaciones6.
Presentamos a continuación el caso clínico de un paciente con manifestaciones atípicas del síndrome de Miller Fisher, su abordaje diagnóstico y manejo terapéutico.
Presentación del casoSe trata de un paciente de 78 años, quien ingresó al servicio de emergencias con un cuadro clínico de 28 horas de evolución caracterizado por desviación de la comisura labial hacia la derecha y oftalmoplejía bilateral, asociado a diplopía, mareo y sensación de debilidad. En la anamnesis no manifestó ningún antecedente patológico o síntomas de infección reciente.
Durante el examen físico se registraron signos vitales en rango de normalidad. En la exploración de las funciones corticales, el paciente se encontraba alerta, orientado globalmente y sin alteraciones en el lenguaje. A la valoración ocular se evidenció ptosis palpebral bilateral, oftalmoplejía generada por la parálisis bilateral de los pares craneales III, IV y VI que se manifestó en forma de un estrabismo divergente de predominio izquierdo. Los reflejos fotomotor y consensual se encontraron indemnes. A nivel facial, se evidenció igualmente desviación de la comisura labial a la derecha con borramiento del surco nasogeniano izquierdo, sin alteraciones en la sensibilidad. Pese a la sensación de debilidad referida por el paciente, no fue evidenciado algún déficit motor o sensitivo en las cuatro extremidades. Los reflejos osteotendinosos fueron simétricos y normales de forma generalizada, la marcha era normal y no se encontraron signos de irritación meníngea. En la auscultación cardiaca y carotídea no se hallaron soplos.
A continuación se realizaron paraclínicos: hemograma, glicemia, proteína C reactiva, TSH, función renal, electrolitos séricos y tiempos de coagulación con reportes dentro de la normalidad; también se realizó una tomografía de cráneo simple que descartó la presencia de lesiones isquémicas, hemorrágicas o desmielinizantes, de instauración aguda o previa. Tras esto, se indicó estancia en sala de hospitalización general, con manejo expectante, para la realización de estudios etiológicos adicionales.
En la revisión del caso en conjunto con el servicio de neurología, se evidenció una progresión de la sintomatología inicial, encontrándose adicionalmente: marcha atáxica e hiporreflexia en los miembros superiores. Como posibles diagnósticos se consideraron la neuropatía craneal múltiple (NCM) y el síndrome de Miller Fisher, por lo que se ordenó la realización de punción lumbar, panel serológico infeccioso, pesquisa imagenológica y paraclínica para neoplasia.
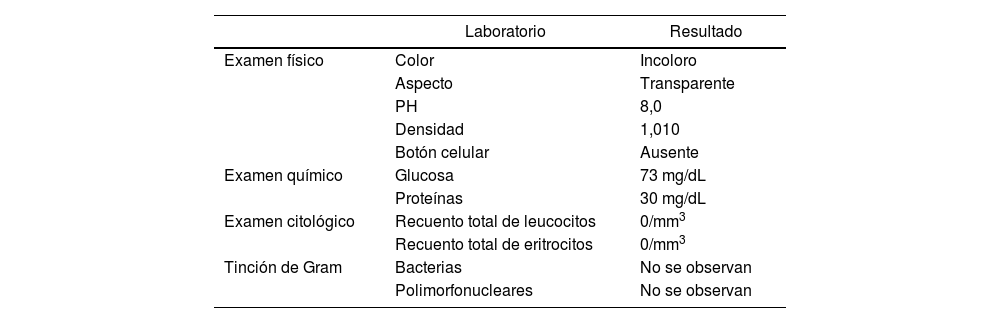
En el examen citoquímico y citológico del líquido cefalorraquídeo no se hallaron alteraciones (tabla 1). La analítica sanguínea, que incluyó el extendido de sangre periférica, reportó una discreta neutrofilia sin alteraciones en las demás líneas celulares. La serología para VIH resultó no reactiva y los niveles séricos de albúmina y calcio colorimétrico se encontraron dentro de los rangos normales. Los resultados de los marcadores tumorales (proteína de Bence Jones y beta-2-microglobulina) fueron negativos; del mismo modo, la electroforesis de proteínas en orina no evidenció resultados patológicos. La tomografía contrastada de tórax, abdomen y pelvis no reveló rastros de neoplasia o sarcoidosis. Por último, se llevó a cabo una resonancia magnética cerebral contrastada que reportó la presencia de pequeños focos de microangiopatía como único hallazgo.
Resultados de muestra de LCR obtenida en punción lumbar
| Laboratorio | Resultado | |
|---|---|---|
| Examen físico | Color | Incoloro |
| Aspecto | Transparente | |
| PH | 8,0 | |
| Densidad | 1,010 | |
| Botón celular | Ausente | |
| Examen químico | Glucosa | 73 mg/dL |
| Proteínas | 30 mg/dL | |
| Examen citológico | Recuento total de leucocitos | 0/mm3 |
| Recuento total de eritrocitos | 0/mm3 | |
| Tinción de Gram | Bacterias | No se observan |
| Polimorfonucleares | No se observan |
Fuente: elaboración propia
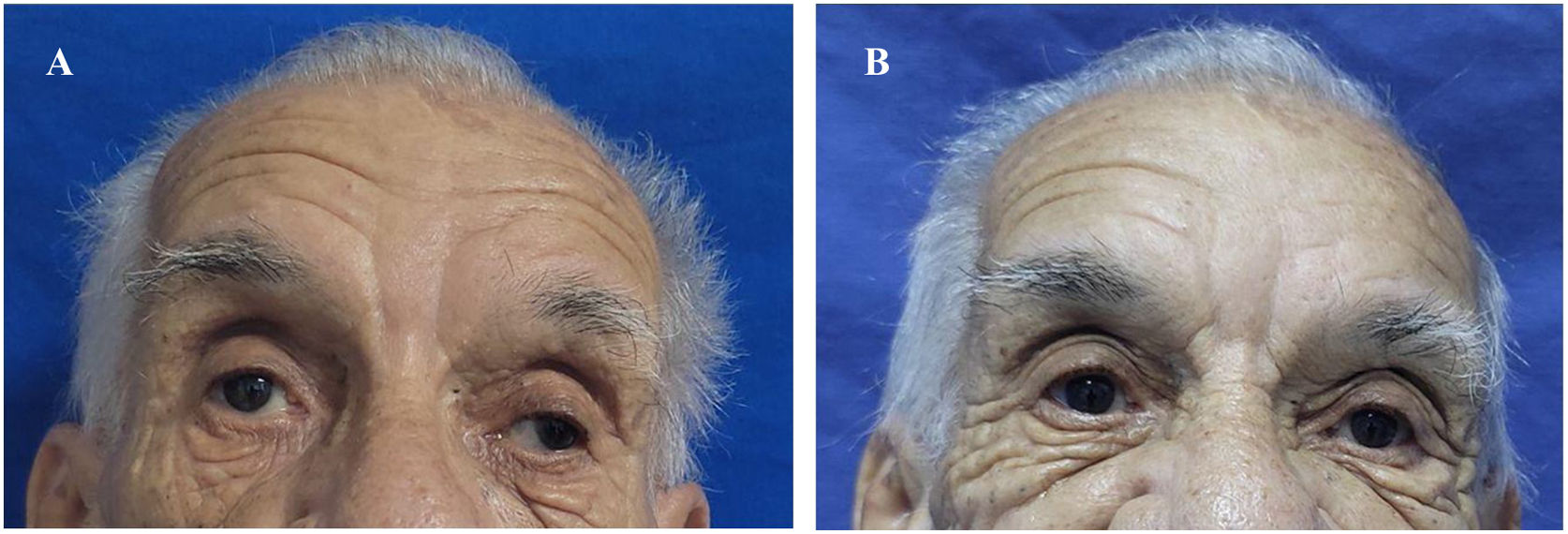
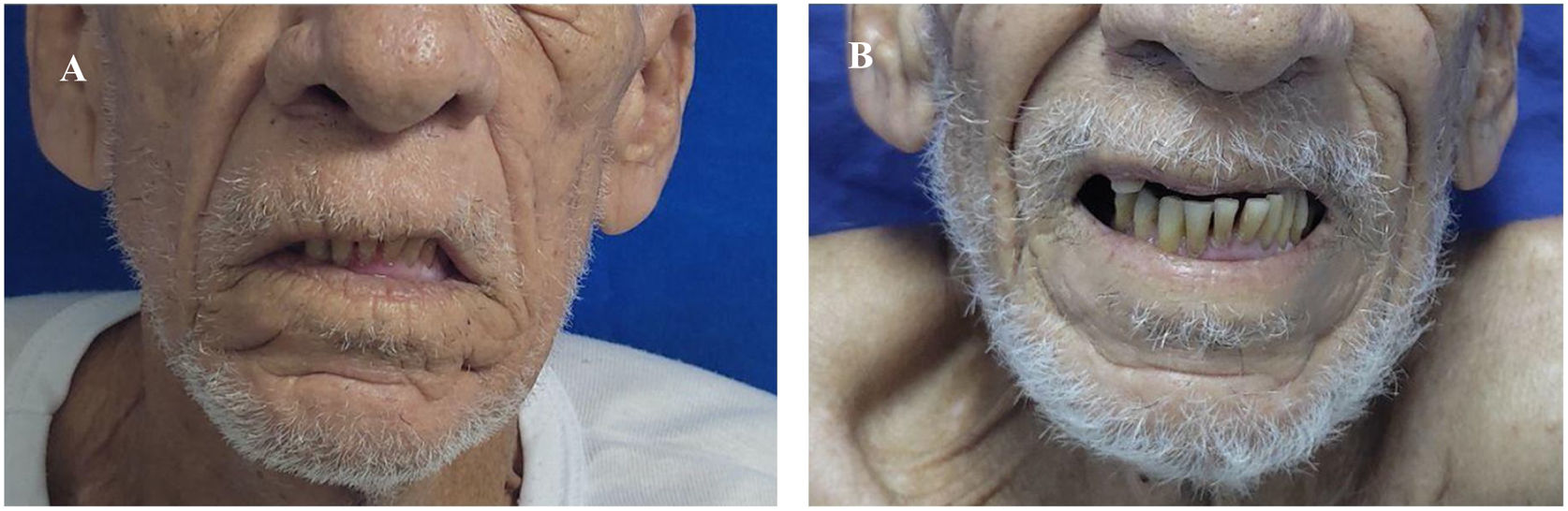
Al descartarse las principales etiologías de la NCM, se hizo diagnóstico de SMF y se indicó traslado a Unidad de Cuidados Intermedios, para la administración de plasmaféresis y monitoreo neurológico. Tras la tercera sesión de plasmaféresis, se observó mejoría de la parálisis facial. Al finalizar sin complicaciones las cinco sesiones programadas, hubo desaparición de la diplopía y del estrabismo; sin embargo, persistía leve parálisis en la mirada horizontal y vertical (fig. 1 y fig. 2). Después de llevar a cabo un monitoreo en sala de hospitalización general y no encontrarse nuevos síntomas neurológicos, se indicó alta médica con órdenes ambulatorias para terapia física y ortóptica, así como seguimiento por los servicios de medicina interna y neurología.

La fotografía A fue tomada antes de iniciar terapia con plasmaféresis. Se realizó exploración de movimientos extraoculares evidenciando ausencia de la mirada conjugada horizontal y vertical. Observamos mirada fija con estrabismo divergente de predominio izquierdo y ptosis palpebral bilateral. La fotografía B fue tomada posterior a cinco sesiones de plasmaféresis. Se obtuvo mejoría del estrabismo y de la parálisis facial, pero con persistencia leve de la parálisis de los músculos extraoculares.

El síndrome de Miller Fisher, una rara enfermedad autoinmunitaria que afecta a los nervios periféricos, se presenta de manera típica con la tríada clásica de oftalmoplejía, ataxia e hiporreflexia. Aun así, otros signos y síntomas neurológicos pueden estar presentes, tales como la diplopía (78%), y otros menos frecuentes como disestesia en las extremidades (14%), ptosis palpebral (2%), parálisis facial (16%), debilidad motora (10%) y alteración de la micción (8%). Las anormalidades pupilares, indicativas de oftalmoparesia interna, también son comunes: asimetría pupilar, reactividad lenta a la luz y disociación cercana a la luz6-7. Al comparar estos hallazgos descritos en la literatura con respecto a lo encontrado en el presente caso clínico, podemos exponer que de forma inicial presentó manifestaciones atípicas tales como la parálisis facial, diplopía y mareo. Simultáneamente, el paciente presentaba la oftalmoplejía como único síntoma de la tríada clásica, por lo que fue un factor confusional que no fue considerado en primera instancia.
Teniendo en cuenta que la sintomatología fue de instauración aguda, se hizo imprescindible descartar otras condiciones más comunes que requieren tratamiento inmediato, como un accidente cerebrovascular, por lo que se llevó a cabo una TAC de cráneo simple, cuyo resultado fue normal.
Posteriormente, durante la evaluación de seguimiento, se encontró marcha atáxica e hiporreflexia en miembros superiores, dato curioso que posibilitó establecer una sospecha diagnóstica de SMF. Aun con la presentación de este signo clínico, por la baja prevalencia de este síndrome, siempre se deben tener en cuenta otras patologías más comunes. Asimismo, dado lo atípico de la presentación inicial, la parálisis de los múltiples pares craneales bilaterales y los nuevos hallazgos neurológicos, resultó de gran importancia considerar la posibilidad de una neuropatía craneal múltiple (NCM). Esta opción cobra gran importancia ya que es fundamental descartar patologías de peor evolución, pronóstico y con mayores complicaciones.
Con este objetivo en mente y para garantizar una mayor eficiencia, fueron puestos en consideración los recursos disponibles en la institución que fueran útiles en la identificación de las principales etiologías de la NCM (tabla 2). Para esto, se realizó punción lumbar, panel serológico infeccioso para ITS, pesquisa imagenológica y paraclínica para neoplasias con TAC toracoabdominopélvico, RMN cerebral, proteína de Bence Jones, beta-2-microglobulina y electroforesis de proteínas. Sin resultados que orientaran a alguna etiología de NCM, se descartó esta como la causa de los síntomas del paciente y se hizo diagnóstico de SMF. Cabe destacar que los estudios del líquido cefalorraquídeo en el SMF reportan comúnmente disociación citoalbuminológica, aunque puede no estar presente al principio de la presentación y puede no desarrollarse en algunos casos6-8, por lo que su ausencia no excluye el síndrome en nuestro paciente.
Etiologías de neuropatía craneal múltiple
| Infecciosas | Bacterianas: enfermedad de Lyme, sífilis, Mycobacterium tuberculosis, Mycoplasma, Brucella y PseudomonasVíricas: VIH, herpes zóster, citomegalovirus, Epstein-BarrFúngicas: criptococosis, histoplasmosis, coccidioidomicosisParasitarias: enfermedad de Chagas, cisticercosis |
| Neoplasias | Tumores primarios del sistema nervioso central, infiltración por linfomas o leucemias |
| Inflamatorias | Sarcoidosis, enfermedad de Behçet, paquimeningitis, lupus eritematoso sistémico, amiloidosis y vasculitis |
| Vasculares | ACV isquémico/hemorrágico troncoencefálico, disección de la arteria carótida |
| Otras | Esclerosis múltiple, encefalitis del tronco del encéfalo, traumatismos y tóxicos |
ACV: accidente cerebrovascular.
Fuente: elaboración propia.
La detección de anticuerpos IgG contra el gangliósido GQ1b, presentes en alrededor del 85% de los pacientes, son el pilar en el diagnóstico paraclínico del SMF; sin embargo, esto constituye una limitante en nuestro caso, debido a la no disponibilidad del laboratorio en nuestra región. De igual forma, pese a múltiples esfuerzos, no fue posible obtener la electromiografía (EMG), debido a pérdida del contacto con el paciente de forma ambulatoria. Esto hubiese sido de mucha utilidad académica, dado que la EMG contribuye a la confirmación diagnóstica, debido a que es posible evidenciar datos objetivos del proceso de desmielinización aguda. Además, el hallazgo de daño axonal se asocia con un peor pronóstico a largo plazo9.
El objetivo del presente artículo es el de reportar una presentación atípica del SMF, el cual representó un reto diagnóstico dado que la sintomatología aguda inicial no fue concluyente para el mismo. Esto nos conduce a destacar la importancia de las evaluaciones seriadas en un paciente con síntomas neurológicos atípicos, llevando a cabo una detallada anamnesis y exploración neurológica, y en general a mantener una postura abierta hacia otras posibles etiologías de acuerdo a la progresión sintomática del paciente, lo que permitirá identificar precozmente esta entidad para la instauración de un tratamiento oportuno, que impacte positivamente en la sintomatología clínica del paciente y su pronóstico a largo plazo.
FinanciaciónNo se recibe financiación.
Conflictos de interésLos autores afirman no tener conflictos de interés a la realización del presente reporte de caso.
Hospital Universidad del Norte, Departamento de Neurología. Atlántico, Colombia.
