






En los últimos años la identificación de anticuerpos y gammapatías monoclonales ha permitido comprender la fisiopatología y favorecer el diagnóstico y tratamiento de una multiplicidad de neuropatías inmunomediadas.
ObjetivoDescribir los anticuerpos de mayor relevancia clínica en las neuropatías, ganglionopatías y neuronopatías inmunomediadas caracterizando en cada caso su valor fisiopatológico o diagnóstico, así como la sensibilidad y especificidad de los métodos utilizados para su determinación.
DesarrolloSe analizarán los anticuerpos identificados en 1) síndrome de Guillain-Barré; 2) polineuropatía inflamatoria desmielinizante crónica (PDIC), 3) neuropatía motora con bloqueo multifocal (NMM); 4) CANOMAD (neuropatía atáxica crónica, oftalmoplejía, proteína IgM monoclonal, aglutininas frías y anticuerpos disialosil); 5) ganglionopatías y neuronopatías y la utilidad de identificar las gammapatías monoclonales.
ConclusionesLos anticuerpos y las gammapatías monoclonales son herramientas que han permitido mejorar el diagnóstico y la comprensión fisiopatológica de las neuropatías inmunomediadas y algunas criptogénicas, así como orientar el tratamiento más adecuado.
Over the last several years the identification of both antineural antibodies and monoclonal gammopathies allowed a better understanding of pathophysiology and improvement in the diagnosis and treatment of many different immune mediated neuropathies.
ObjectiveTo describe the antineural antibodies of greater clinical utility in the diagnosis of immune mediated neuropathies and neuronopathies. In each case we underline their value in either the pathophysiology or diagnosis of these disorders as well as the sensitivity and specificity of the diagnostic techniques currently in use.
DevelopmentWe will review the antibodies identified in 1) Guillain-Barré syndrome; 2) Chronic inflammatory demyelinating polyneuropathy (CIDP); 3) Multifocal motor neuropathy (MMN); 4) Chronic Ataxic Neuropathy Ophthalmoplegia M-protein Agglutination Disialosyl antibodies syndrome (CANOMAD); 5) Ganglionopathies and Neuropathies and the value of identifying monoclonal gammopathies.
ConclusionsThe antibodies and monoclonal gammopathies are useful tools in both the diagnosis and understanding of the mechanisms involved in immune mediated and cryptogenic neuropathies and orienting treatment.
El sistema inmune es capaz de diferenciar entre componentes de nuestro organismo y moléculas extrañas, siendo una de sus principales funciones evitar los efectos deletéreos de agentes patógenos. Sin embargo, en algunos huéspedes predispuestos, esta actividad, al principio protectora, puede desencadenar una respuesta inmunitaria exagerada contra antígenos propios y generar una enfermedad autoinmune. Los procesos autoinmunes requieren de la combinación de una predisposición genética, la presencia de factores ambientales desencadenantes o condicionantes y la desregulación inmunitaria con pérdida de los mecanismos de tolerancia1–3.
En el sistema nervioso periférico la barrera hemato-nerviosa mantiene separado al sistema inmunitario. En algunas circunstancias la barrera se interrumpe y factores tanto humorales como celulares pueden acceder y originar enfermedades neuromusculares inmunológicamente mediadas1. Estas enfermedades comprenden los trastornos del nervio, unión neuromuscular y músculo2.
La identificación de los anticuerpos dirigidos contra el receptor nicotínico de acetilcolina (AChRA) lideró el camino en la descripción de una amplia variedad de autoanticuerpos que resultan útiles para el diagnóstico, clasificación y pronóstico de estos desórdenes2,4,5. La descripción posterior de anticuerpos tales como anti-tirosina quinasa específica de músculo (anti-MuSK), anticuerpos contra estructuras nodales, paranodales y anticuerpos antisintetasa fue crítica para la detección de distintos fenotipos dentro de los síndromes clínicos clásicos2,5,6.
Los anticuerpos son inmunoglobulinas, generalmente del tipo IgG, que pueden estar involucradas directamente en la fisiopatología o solo ser marcadores con una importancia incierta en la patogénesis. Un anticuerpo tiene actividad patogénica si cumple con los criterios de unión a estructuras específicas produciendo cambios patognomónicos, inducción de tales cambios en cultivo celular y transferencia de enfermedad a animales de experimentación. En general, solo una proporción es realmente patogénica, por lo que en muchas ocasiones el título del anticuerpo no se correlaciona con la severidad de la enfermedad. Otros factores tales como especificidad del epítope e isotipo contribuyen a la capacidad patogénica de cada autoanticuerpo1,3.
Las pruebas serológicas para la determinación de anticuerpos se han expandido en función del descubrimiento de las etiologías autoinmunes2,5.
El presente trabajo provee una revisión de las neuropatías inmunomediadas haciendo foco en el anticuerpo y fenotipo asociado, metodología para la determinación y su utilidad en la práctica clínica habitual.
MétodosSe consultaron las bases de datos PubMed-NCBI y Lilacs utilizando las palabras: anticuerpos y Guillain-Barré, CIDP, neuropatía motora multifocal, CANOMAD, ganglionopatía, neuronopatía y gammapatía monoclonal.
Se analizó en cada caso la historia de la identificación del anticuerpo, su modo de detección y el valor clínico de los mismos.
Detección de los anticuerposLos métodos básicos para la detección de anticuerpos en suero son el radioinmunoanálisis (RIA), el ensayo de inmuno-absorción ligado a enzima (ELISA), inmunofluorescencia indirecta (IFI), Western blot, ensayos celulares. El RIA utiliza antígeno purificado parcialmente marcado con material radioactivo. El suero del paciente se mezcla con el antígeno marcado y se adicionan anticuerpos dirigidos a anticuerpos humanos, los cuales provocan la precipitación de los complejos antígeno marcado-anticuerpo. El precipitado es sometido a un contador de radioactividad, lo cual permite la cuantificación de anticuerpos séricos dirigidos contra el antígeno objetivo3. El método ELISA implica inmovilización del antígeno en un plato de microtitulación al que se adiciona el suero del paciente permitiendo la unión, y luego se agrega un anticuerpo secundario que reconoce al anticuerpo del paciente asociado con una enzima. Finalmente, el agregado de un sustrato para dicha enzima lleva a la producción de un precipitado coloreado. Una dilución serial del suero del paciente provee el título de anticuerpos (mayor dilución, mayor concentración de anticuerpos)3. La IFI requiere cultivo celular o una fracción de tejido como fuente de antígeno. La determinación del antígeno antinuclear es un ejemplo de esta metodología. El suero del paciente es adicionado a líneas celulares epiteliales humanas. Luego se agregan anticuerpos marcados con fluorescencia que se unen a anticuerpos humanos. La preparación es observada con microscopio de fluorescencia. El patrón de tinción y la dilución en la cual la unión es apreciada son informados en el resultado3.
El método Western blot comprende el aislamiento de muestras proteicas y se basa en la separación en función del peso molecular en un gel de electroforesis. Las proteínas en gel son transferidas a una membrana. Se agrega un anticuerpo específico dirigido a la proteína bajo estudio. Luego de la incubación, un segundo anticuerpo es agregado para identificar el primero, generalmente asociado a una enzima para cuantificar la reacción3. En el ensayo con cultivos celulares, la prueba celular detecta anticuerpos dirigidos contra, por ejemplo, AChR «agrupados». Mediante este ensayo celular se detectan anticuerpos dirigidos contra los AChR agrupados en aproximadamente el 50% de los individuos miasténicos con AChRA RIA negativo4,5.
Anticuerpos antigangliósidos y síndrome de Guillain-BarréEl síndrome de Guillain-Barré (SGB) es una polirradiculoneuropatía de etiología autoinmune postinfecciosa y de curso monofásico caracterizada por la presencia de debilidad muscular simétrica de instalación aguda (4 semanas) asociada a arreflexia y trastornos sensitivos distales en las extremidades7. Es frecuente el compromiso de pares craneales y en el 20% de los pacientes el cuadro evoluciona a insuficiencia respiratoria. Se han descripto diferentes variantes (tabla 1), la más frecuente es la llamada polirradiculoneuropatía desmielinizante inflamatoria aguda (AIDP, por sus siglas en inglés). Su incidencia es de 0,6 a 4 casos por 100.000 por año en todo el mundo8. Existen variantes axonales asociadas a anticuerpos antigangliósidos y el síndrome de Miller-Fisher, caracterizado por oftalmoplejía, ataxia y arreflexia.
Variantes clínicas del síndrome de Guillain-Barré y los anticuerpos asociados más frecuentemente
| Variante clínica | Anticuerpos asociados |
|---|---|
| Polirradiculoneuropatía inflamatoria desmielinizante aguda (AIDP en inglés) | Desconocido |
| Neuropatía axonal motora y sensitiva (AMSAN en inglés) | GM1, GM1b, GD1a |
| Neuropatía axonal motora aguda (AMAN en inglés) | GM1, GM1b, GD1a, GalNac-GD1a |
| Neuropatía sensitiva aguda | GD1b |
| Variantes focales | |
| Síndrome de Miller-Fisher | GQ1b, GT1a |
| Variante faringobraquial | GT1a |
| Síndromes de superposición | |
| Miller-Fisher/Guillain-Barré | GQ1b, GM1, GM1b, GD1a, GalNac-GD1a |
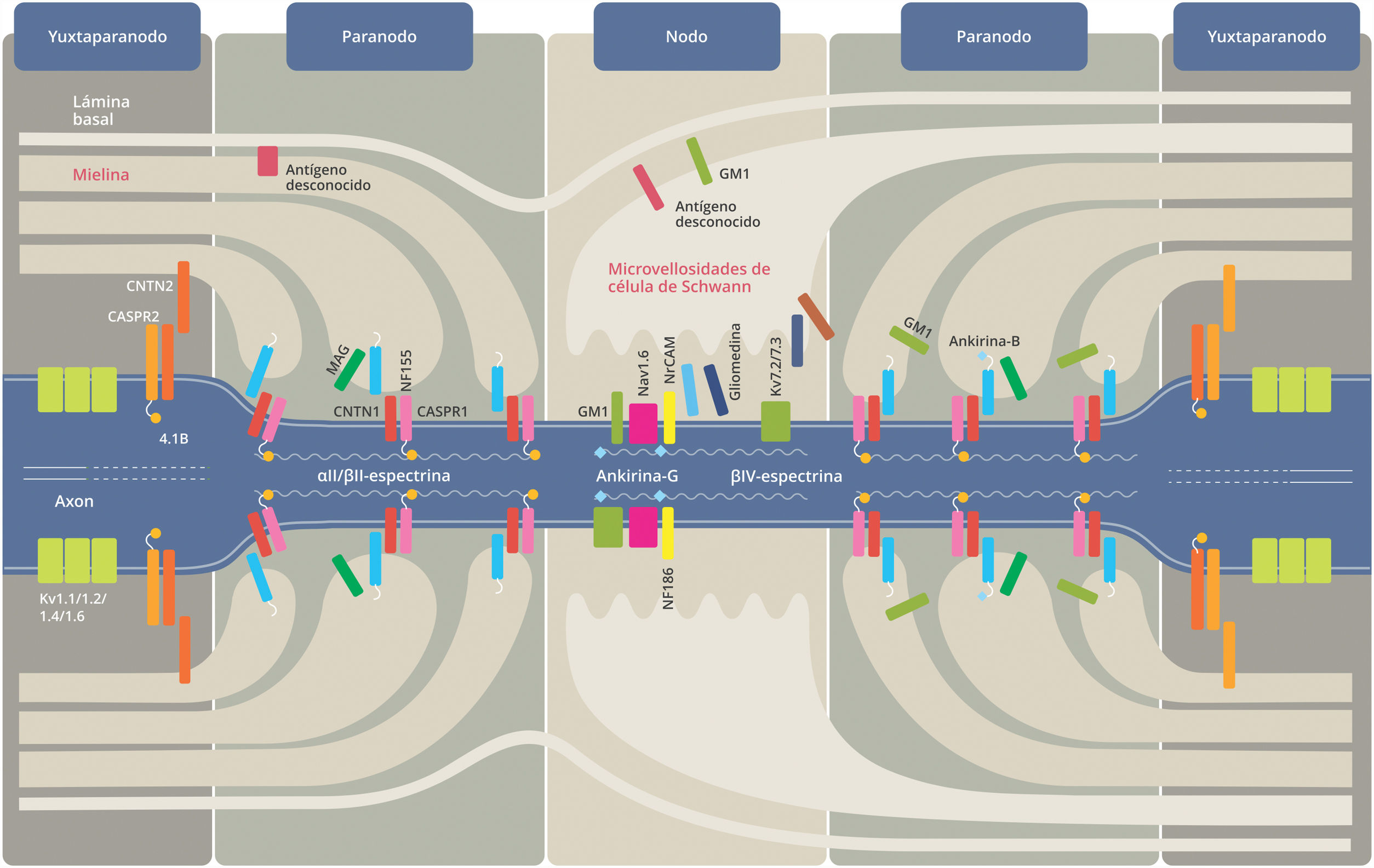
Los mecanismos involucrados en la AIDP no están del todo dilucidados, se desconocen los antígenos asociados a la mielina de los nervios periféricos que median respuestas autoinmunes. En las formas axonales el ataque autoinmunitario está dirigido hacia la porción oligosacárida de glicolípidos de la membrana axonal denominados gangliósidos. La estructura química de los oligosacáridos expresados en la membrana de los microorganismos es muy similar a la de los gangliósidos humanos, por lo que este mecanismo de «mimetismo molecular» estimula a células B a producir anticuerpos que se fijan sobre estos gangliósidos ubicados en la periferia de los nodos de Ranvier9 (fig. 1). Esto genera activación de complemento, formación del complejo de ataque de membrana y la disrupción de los canales de sodio dependientes de voltaje ubicados en el nodo10. La pérdida de la arquitectura normal del nodo de Ranvier hace que la conducción nerviosa se vea afectada con la consiguiente aparición de bloqueos de conducción11 y debilidad muscular, así como la entrada de macrófagos al espacio periaxonal, dañando los axones.

Nodo de Ranvier y su estructura.
CASPR: proteína asociada a la contactina; CNTN: contactina; Kv: canales de potasio dependientes de voltaje; MAG: glucoproteína asociada a la mielina; Nav: canales de sodio dependientes de voltaje; NF: neurofascina; NrCAM: molécula de adhesión celular neuronal.
Modificado de Querol et al.31.
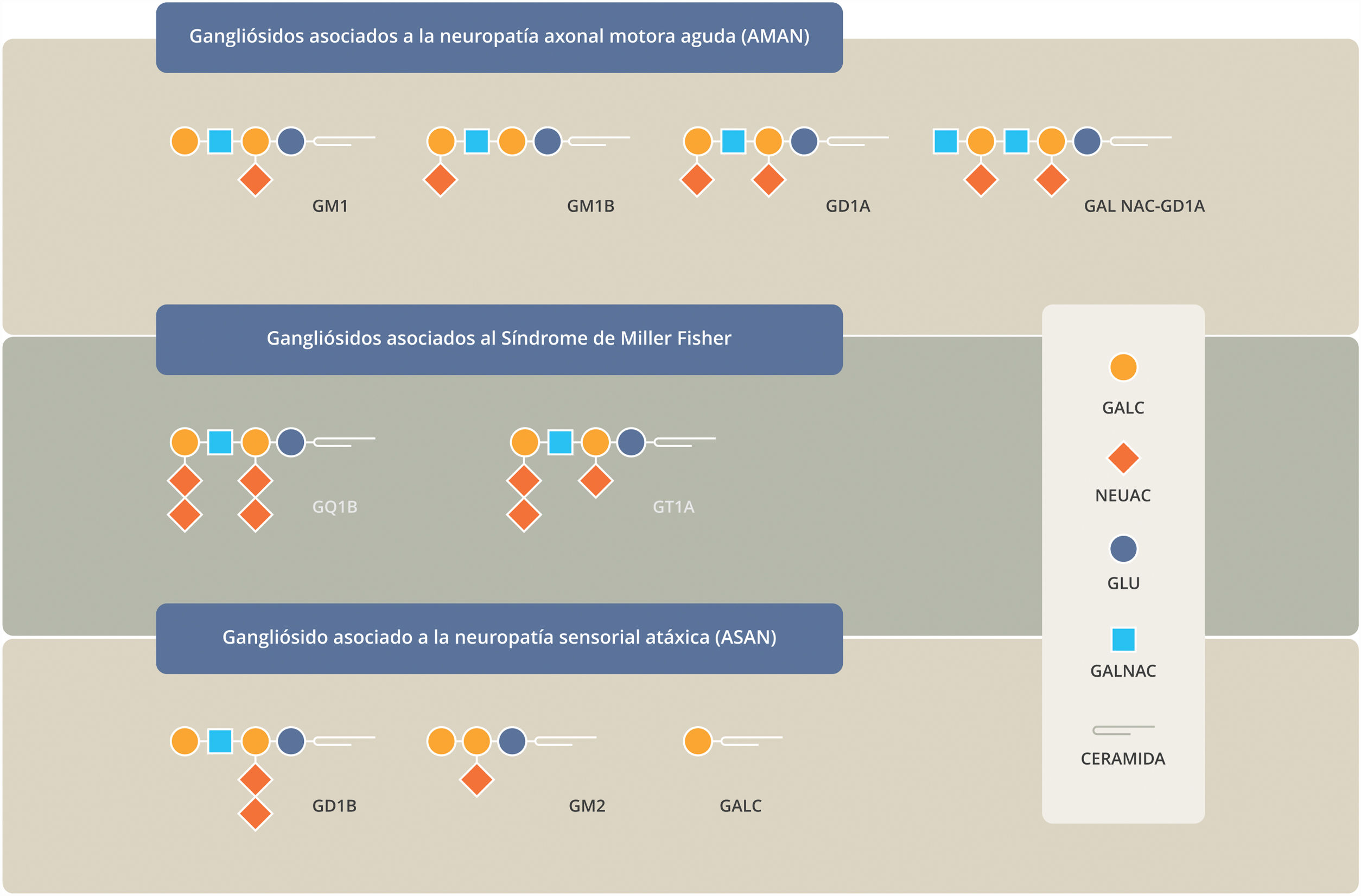
La primera descripción de la presencia de anticuerpos circulantes contra glucoesfingolípidos (gangliósidos) en pacientes con SGB fue en 1988 en una serie de casos con 26 pacientes, de los cuales 5 presentaban respuesta de tipo IgG contra LM1 y su hexosa analógica Hex-LM1, 2 contra GD1b (IgG) y 2 más contra GD1a y GT1b (IgM)12. Los títulos de anticuerpos en estos casos disminuyeron con la mejoría clínica. Los glucoesfingolípidos están compuestos por una ceramida (N-aciladaesfingosina) unida a uno o más azúcares (hexosas) y a un número variable de moléculas de ácido siálico13 (fig. 2). La ceramida hidrófoba está inmersa en la membrana lipídica y la estructura hidrofílica de carbohidratos se expone extracelularmente, en las membranas plasmáticas, actuando como antígeno. Los gangliósidos más conocidos son GM1, GD1a, GD1b y GT1b. Pertenecen a la serie G1, donde G significa gangliósido. Los 4 principales gangliósidos difieren con respecto al número y la posición de sus ácidos siálicos, donde M, D y T representan mono-, di- y grupos tri-sialosilo. Los gangliósidos tienen una distribución extensa en todos los tejidos predominando en el sistema nervioso, particularmente en los paranodos, en las terminales presinápticas del axolema y en las regiones adaxonal y axolema internodal14. Se ubican en la cara externa de las membranas celulares, por lo que carecen de respuesta intracelular directa. El isotipo IgG es el asociado a las diferentes variantes de SGB. Las asociaciones clínico-serológicas demostradas se resumen en la tabla 1. Los anticuerpos GM1b (IgG) tienen una asociación más frecuente entre la infección precedente con Campylobacter jejuni y la variante axonal (AMAN). Presentan un curso más rápido, severo, con debilidad distal predominante y con recuperación más lenta. Los anticuerpos anti-GQ1b se asocian al síndrome de Miller-Fisher y a la encefalitis de Bickerstaff7,15,16.
. GALC: galactosa; NEUAC: ácido N-acetil neuroamínico; GALNAC: N-acetilgalactosamina; GLU: glucosa. Modificado de Willison y Yuki13.")
Estructura molecular de gangliósidos y galactocerebrósidos (GalC).
GALC: galactosa; NEUAC: ácido N-acetil neuroamínico; GALNAC: N-acetilgalactosamina; GLU: glucosa.
Modificado de Willison y Yuki13.
ELISA: los títulos de anticuerpos se expresan como relaciones porcentuales con respecto a un calibrador y se asignan a distintas categorías de título (negativo, zona gris, positivo y fuertemente positivo).
Inmunoblot (Euroimmun): esta técnica utiliza tiras reactivas que contienen fracciones de gangliósidos purificados que son utilizados como substratos. Los autoanticuerpos se unen inicialmente a su antígeno específico y después son detectados por un revelado colorimétrico mediante la reacción con un antígeno secundario acoplado a una enzima. La tinción de tiras de inmunoblot se escanean posteriormente y se analizan con un software especial de Euroimmun.
Sensibilidad y especificidad del métodoELISA (Kit GanglioCombiBuhlmann)>50: positivo. IgG: sensibilidad 32%, especificidad 97%.
Inmunoblot (Euroimmune): valores ≥+1 se consideran positivos.
IgG: sensibilidad 56%, especificidad 100%. IgG+IgM: sensibilidad 98%, especificidad 60%17.
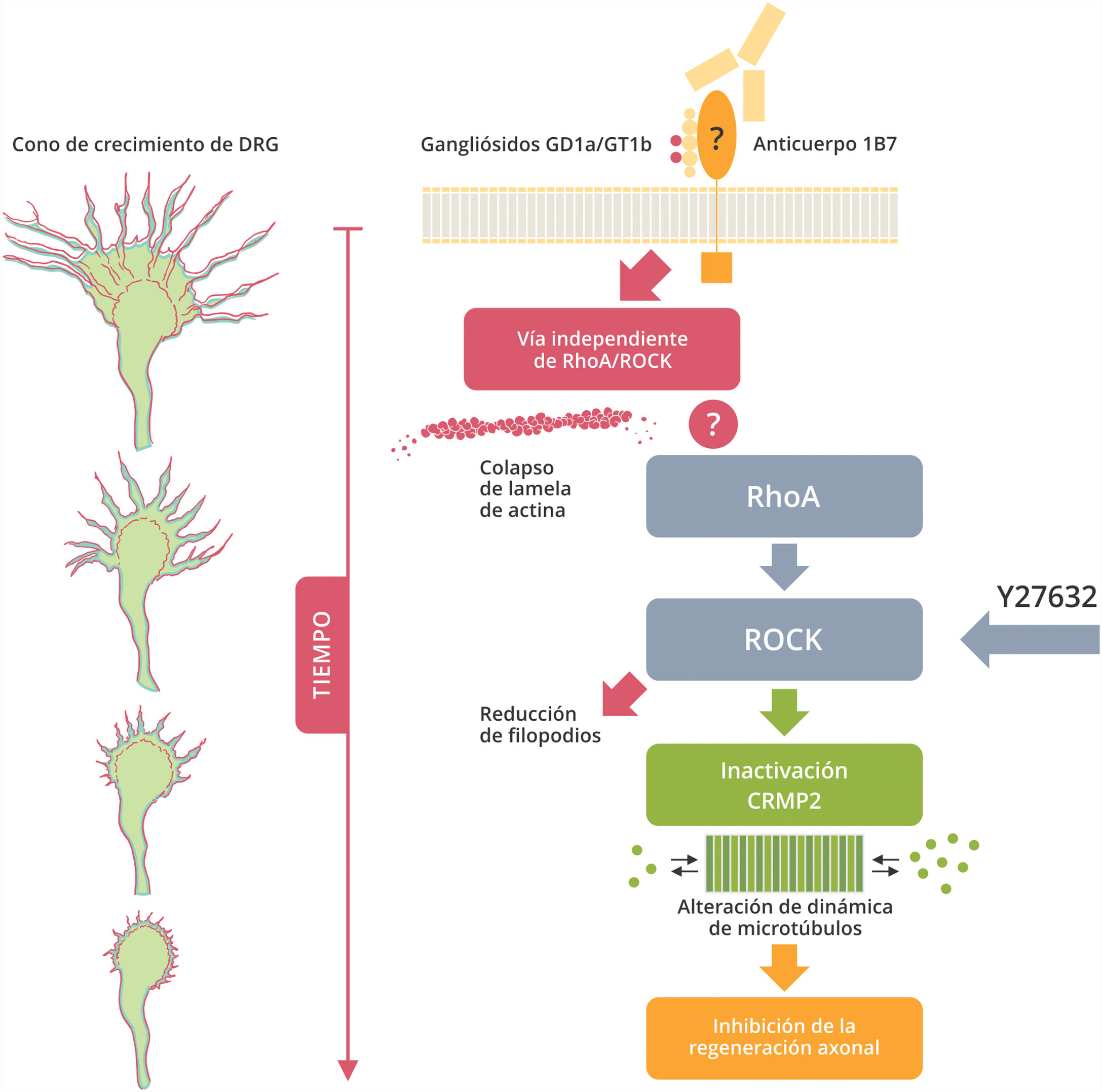
Autoinmunidad y regeneración nerviosaHoy sabemos que en el SBG el espectro clínico de recuperación varía considerablemente entre los pacientes, en los cuales hasta un 20% presentan severas dificultades motoras al año de haber padecido la enfermedad18. Esto denota la existencia de diversos factores que condicionan el daño nervioso y su capacidad de regeneración19. Uno de los factores más estudiados fue precisamente la relación de los anticuerpos antigangliósidos con la severidad de la enfermedad y su pronóstico. Numerosos trabajos demostraron la vinculación entre la presencia de anticuerpos antigangliósidos circulantes y una recuperación clínica incompleta en pacientes con variantes clínicas tanto axonales como desmielinizantes20–24. Trabajos experimentales demostraron el rol inhibitorio de los anticuerpos antigangliósidos en la regeneración axonal. Dicha inhibición se correlacionó con la presencia de estructuras desorganizadas en su conformación microtubular denominadas conos de crecimiento distróficos en el área de lesión poniendo de manifiesto intentos fallidos de regeneración axonal posterior a un daño nervioso25,26. Estudios posteriores, tanto in vitro como in vivo, establecieron las primeras asociaciones entre la inhibición del crecimiento neurítico con anticuerpos antigangliósidos y la activación consiguiente de la pequeña GTPasa RhoA (Ras homolog gene family, member A, en inglés) y su quinasa asociada ROCK27–30 (Rho-associated protein kinase, en inglés). El bloqueo farmacológico selectivo de esta vía intracelular podría constituir un potencial blanco terapéutico a fin de estimular la regeneración axonal en aquellas variantes de SGB asociadas a fallas en la regeneración (fig. 3).
![Mecanismos intracelulares gatillados por la unión del anticuerpo 1B7 (análogo murino GD1a/GT1b) con el receptor de tipo gangliósido. La unión genera una transducción de señal que activa 2 vías diferentes, una dependiente y otra independiente de RhoA (Ras homolog gene family, member A, en inglés). La vía de RhoA y su efector ROCK (Rho-associated protein kinase, en inglés) genera inhibición de la regeneración axonal por interacción con proteínas vinculadas a la estabilización de microtúbulos (Collapsin response mediator protein [CRMP2], en inglés, entre otras). Diferentes fármacos son capaces de bloquear selectivamente esta vía a diferentes niveles (p.ej., Y-27632) constituyendo un potencial blanco terapéutico.](https://static.elsevier.es/multimedia/18530028/0000001200000002/v1_202006160636/S1853002820300185/v1_202006160636/es/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9ikrjRGWJ7XO1V/0BzprPABjGVq2kNLIZRepF47Jt5bffk5hlKKNZ7yHGFlQ7GJCTITEdEEMnSVvJZLigkcDT/dStAKrEKOHS3cdEwHQbiPBfvlvQ5QkD1PsHogjjc8/7yD3BzuB8n1gV4qmCOT7HOEkhPCIBTiyO+0JvPNkgq68SAl1OMpWZ47FEwD3zlhox4gjYrw3IjamhVd5W7rVN46EDxI/znV4p1mRflRUTYn+k= "Mecanismos intracelulares gatillados por la unión del anticuerpo 1B7 (análogo murino GD1a/GT1b) con el receptor de tipo gangliósido. La unión genera una transducción de señal que activa 2 vías diferentes, una dependiente y otra independiente de RhoA (Ras homolog gene family, member A, en inglés). La vía de RhoA y su efector ROCK (Rho-associated protein kinase, en inglés) genera inhibición de la regeneración axonal por interacción con proteínas vinculadas a la estabilización de microtúbulos (Collapsin response mediator protein [CRMP2], en inglés, entre otras). Diferentes fármacos son capaces de bloquear selectivamente esta vía a diferentes niveles (p.ej., Y-27632) constituyendo un potencial blanco terapéutico.")
Mecanismos intracelulares gatillados por la unión del anticuerpo 1B7 (análogo murino GD1a/GT1b) con el receptor de tipo gangliósido.
La unión genera una transducción de señal que activa 2 vías diferentes, una dependiente y otra independiente de RhoA (Ras homolog gene family, member A, en inglés). La vía de RhoA y su efector ROCK (Rho-associated protein kinase, en inglés) genera inhibición de la regeneración axonal por interacción con proteínas vinculadas a la estabilización de microtúbulos (Collapsin response mediator protein [CRMP2], en inglés, entre otras). Diferentes fármacos son capaces de bloquear selectivamente esta vía a diferentes niveles (p.ej., Y-27632) constituyendo un potencial blanco terapéutico.
Los anticuerpos antiglicolípidos han sido esenciales en la comprensión de la fisiopatología y el desarrollo de diferentes variantes clínicas del SGB. Títulos elevados y persistentes podrían estar asociados a un peor pronóstico de la enfermedad. Sin embargo, su valor diagnóstico es limitado y dependiente de la metodología utilizada para su detección. La presencia de los mismos en cuadros clínicos dudosos ayuda a confirmar el diagnóstico, pero un estudio negativo jamás lo descarta. No se debe dilatar el inicio del tratamiento a la espera de estos anticuerpos.
Anticuerpos en neuropatías inmunomediadas crónicas: polineuropatía inflamatoria desmielinizante crónica (PDIC) y neuropatía motora multifocal (NMM)Polineuropatía inflamatoria desmielinizante crónica (PIDC)La polineuropatía inflamatoria desmielinizante crónica (PIDC) tiene una prevalencia de entre 1 y 9 casos por cada 100.000 habitantes31–34. Clásicamente presenta debilidad simétrica proximal y distal, alteraciones sensitivas y arreflexia, que se desarrollan durante un período mínimo de 8 semanas. La PIDC es una enfermedad heterogénea, con múltiples fenotipos, cuya fisiopatología es mayormente desconocida. La respuesta terapéutica a inmunoglobulina endovenosa (IGEV), plasmaféresis y corticoides, así como estudios de transferencia pasiva en modelos animales35, sugieren la presencia de factores inmunes humorales y celulares involucrados en su fisiopatología. La presencia de inmunoglobulinas y depósitos de complemento en biopsias de nervio de pacientes con PIDC también apoyan la teoría de un origen autoinmune36. Un estudio reciente demuestra que un pequeño subgrupo de pacientes con anticuerpos dirigidos contra estructuras paranodales presentan en biopsias de nervio sural desprendimiento de mielina en las uniones axo-gliales del paranodo asociado a degeneración axonal, sin observarse la clásica desmielinización por infiltración macrofágica, sugiriendo multiplicidad de mecanismos patogénicos37.
El diagnóstico de PIDC se basa en criterios clínicos, electrofisiológicos y de laboratorio. La guía conjunta de la Federación Europea de Sociedades Neurológicas y la Sociedad de Nervio Periférico (EFNS/PNS) reúne los criterios más utilizados por su alta sensibilidad y especificidad para el diagnóstico38.
Diferentes autoanticuerpos contra antígenos mielínicos (proteína mielínica zero, proteína de la mielina periférica 2 y 22, conexina, proteína asociada a la mielina: MAG) han sido descriptos en pacientes con PIDC, pero no fueron identificados en la mayoría de los pacientes, no han sido confirmados en grandes cohortes, ni su medición está incluida en los criterios diagnósticos vigentes. La investigación en los últimos 10 años ha demostrado la existencia de una serie de anticuerpos dirigidos a estructuras del nodo de Ranvier con relevancia tanto clínica como terapéutica de la PIDC39,40. Las regiones nodales, paranodales y yuxtaparanodales son sitios de localización de importantes canales iónicos, proteínas estructurales y moléculas de adhesión con dominios extracelulares accesibles a diferentes anticuerpos41. Las proteínas de la unión axo-glial cumplen un rol fundamental de anclaje entre el axón y la mielina a nivel paranodal, en la formación y mantenimiento del nodo de Ranvier y en la organización de los canales iónicos42 (fig. 1). Anticuerpos del tipo IgG4 contra los antígenos paranodales neurofascina-155 y contactina-1 han sido consistentemente descriptos en un pequeño subgrupo de pacientes con PIDC que presentaron características clínicas específicas y, en especial, escasa mejoría con IGEV43,44. Entre el 6 y 18% de los pacientes con PDIC presentan anticuerpos contra neurofascina-155 que son altamente específicos, se caracterizan por ataxia sensitiva y/o cerebelosa, temblor discapacitante, menor edad, evolución aguda-subaguda y debilidad distal con pobre respuesta terapéutica a la IGEV pero buena a rituximab43,45. En algunos casos combinan desmielinización central (remedando las lesiones de la esclerosis múltiple pero con bandas oligoclonales negativas)46. Los anticuerpos contra contactina-1 se detectan en el 8% de los pacientes asociados a una edad más avanzada, debilidad de presentación subaguda y agresiva de inicio (símil SGB), temblor no tan frecuente, compromiso axonal precoz y también escasa respuesta a IGEV, pero buena a corticoides y rituximab44. El reconocimiento de estos anticuerpos ha permitido establecer una correlación inmunológica-clínica y ha demostrado tener implicancias diagnósticas y pronósticas47. Otros anticuerpos nodales y paranodales han sido descriptos en publicaciones aisladas y en un muy bajo número de pacientes. Este es el caso de la proteína asociada a la contactina (CASPR-1), antígeno paranodal descripto en un paciente con PIDC y otro con SGB. Ambos pacientes tenían intenso dolor neuropático31,48 y el isotipo de Ig identificado fue IgG3 en el paciente con SGB e IgG4 en el que tenía PIDC (fig. 1). Anticuerpos dirigidos contra isoformas nodales de neurofascina tanto 186 como 140 también han sido identificados. Los portadores presentaron inicio subagudo, ataxia sensitiva, compromiso de pares craneales y bloqueos de conducción; además, uno de ellos desarrolló glomeruloesclerosis y otro fibrosis retroperitoneal, reforzando de esta manera el papel patogénico de estos anticuerpos IgG440 (fig. 1). Otros anticuerpos de relevancia no específica en PIDC con reactividad contra gliomedina, moesina o contactina asociada a la proteína 2 (CASPR-2) han sido descriptos40.
Conclusión anticuerpos en PIDCLos anticuerpos nodales y paranodales se identifican solamente en un pequeño subgrupo de pacientes con PIDC. Mejoran la comprensión de la fisiopatología, definen fenotipos clínicos y ayudan a seleccionar terapias. Su determinación por el momento no está comercialmente disponible, y solo puede realizarse en laboratorios de referencia en el exterior.
Neuropatía motora multifocalLa neuropatía motora multifocal (NMM) constituye una forma de neuropatía que afecta preferentemente a jóvenes, cursa con debilidad muscular lentamente progresiva de distribución asimétrica o focal en el territorio de 2 o más nervios, especialmente en los miembros superiores, y ausencia tanto de síntomas sensitivos como de signos de compromiso de la neurona motora superior. En algunos casos calambres y fasciculaciones pueden acompañar al cuadro, y el hallazgo de bloqueos de la conducción nerviosa es su rasgo esencial en los estudios neurofisiológicos. Si bien la fisiopatología no es del todo conocida, su respuesta favorable a la terapia con IGEV y su relación con la detección en sangre de anticuerpos hacen suponer el origen inmunomediado49. La asociación entre la NMM con niveles séricos elevados de anticuerpos IgM contra GM1 fue descripta por primera vez en 1988 por Pestronk et al., junto con la respuesta efectiva a la inmunoterapia50.
El antígeno GM1 se expresa de forma ubicua, pero se observa con mayor concentración en los nervios motores, en los nodos de Ranvier y en los paranodos adyacentes11 (fig. 1). Se considera fundamental para la estabilización paranodal, el agrupamiento de canales iónicos y el mantenimiento de la conducción del potencial de acción51,52. Los títulos del anticuerpo se relacionaron con los depósitos de complemento de manera directamente proporcional, y dichos depósitos se redujeron mediante la utilización de IGEV de forma dosis dependiente53. Esto sugiere el rol fundamental del complemento en la cascada inmunológica de la NMM.
La prevalencia de anticuerpos IgM anti-GM1 en pacientes con NMM varía de acuerdo a las series publicadas, oscilando de un 20 a un 85%, siendo del 43% en un estudio reciente de 88 pacientes con NMM utilizando la metodología ELISA54. Esta variabilidad se debe en parte a que no existe un consenso en la técnica de detección, y a la falta de un método gold standard para la medición de los anticuerpos. La especificidad de la detección de anticuerpos anti-GM1 en NMM es baja, ya que estos anticuerpos también son detectados en un 10% de pacientes con esclerosis lateral amiotrófica, otras enfermedades neurológicas y aun en controles sanos. La detección de complejos heteroméricos de anticuerpos IgM anti-GM1 y anti-galactocerebrósido (GM1: GalC) aumenta la sensibilidad diagnóstica del método en la NMM, utilizando tanto la técnica de ELISA como la de glicoarray55.
El único estudio que ha reportado una relación entre el anticuerpo detectado y la clínica del paciente señala que aquellos que presentan reactividad para IgM anti-GM1 presentan mayor debilidad, mayor discapacidad y mayor pérdida axonal, en comparación con pacientes anti-GM1 seronegativos. El mismo estudio demuestra correlación positiva entre los títulos de anticuerpos IgM anti-GM1 y el puntaje de la escala Medical Research Council56. Otros anticuerpos menos frecuentemente reportados incluyen: asialo-GM1, GD1A y GM211,57. Anticuerpos contra estructuras nodales y paranodales encontrados en PICD, como neurofascina-155, contactina-1, neurofascina-186 y gliomedina, no han sido encontrados en NMM.
Conclusión anticuerpos en NMMLa detección de anticuerpos anti-GM1 puede ser una prueba de soportediagnóstico útil en la NMM55,58. Sin embargo, el cuadro clínico y el neurofisiológico son esenciales y el anticuerpo no puede de forma aislada confirmar o descartar el diagnóstico de esta enfermedad.
Neuropatías y paraproteínasEl término paraproteinemia alude al desorden a partir del cual poblaciones clonales de linfocitos B o células plasmáticas producen y secretan proteínas en exceso. Estas proteínas monoclonales (proteína M) son inmunoglobulinas compuestas por 2 cadenas pesadas y 2 cadenas livianas (principalmente IgM, IgG e IgA) o constituidas por cadenas livianas libres (kappa o lambda)59.
El proceso proliferativo que da origen a la paraproteinemia puede ser premaligno, como es el caso de la gammapatía monoclonal de significado incierto (GMSI); o tratarse de un desorden maligno sistémico, como el mieloma múltiple (MM), la macroglobulinemia de Waldenstrom (MW), o el síndrome POEMS (polineuropatía-organomegalia-endocrinopatía-proteínaM-alteraciones dérmicas) o la amiloidosis primaria (AL)60.
Gammapatía monoclonal de significado inciertoDos terceras partes de los casos de paraproteinemia se asocian a GMSI. Sus características incluyen: nivel de proteínaM menor de 30g/L, menos del 10% de células plasmáticas en médula ósea, ausencia o niveles bajos de proteínaM en orina y ausencia de otros elementos sistémicos como anemia, hipercalcemia, lesiones óseas o falla renal. La progresión a malignidad es la principal consecuencia clínica de GMSI y ocurre con una frecuencia del 1% anual. Si se trata de GMSI IgM, el riesgo es la transformación a MW, mientras que si se trata de GMSI IgA o IgG la conversión es hacia MM61.
La prevalencia de GMSI aumenta con la edad: es del 3% en mayores de 50 años y del 7,5% en mayores de 85 años62.
Macroglobulinemia de WaldestromLa MW es un desorden linfoproliferativo de células B, caracterizado por infiltración linfoplasmocitaria en la médula ósea y gammapatía monoclonal (GM) IgM en sangre. Típicamente se presenta en la 7.a década de la vida, con predominancia masculina (2:1). Su clínica incluye hepatoesplenomegalia, linfadenopatías e hiperviscosidad60–63.
La neuropatía es usualmente distal simétrica, sensitiva más que motriz y desmielinizante, similar a la observada en pacientes con GMSI IgM60.
Mieloma múltipleEl MM es la neoplasia hematológica más comúnmente asociada a paraproteinemia. La edad media de inicio es 65 años. En el 90% de los pacientes se detecta proteínaM sérica, más comúnmente IgG, seguida por IgA. En el 20% de los casos se detectan solo cadenas livianas monoclonales. El diagnóstico de MM se basa en la presencia de proteínaM en suero, proteinuria de Bence-Jones, >10% de células plasmáticas en médula ósea y evidencia de daño de órgano blanco, como hipercalcemia, insuficiencia renal, anemia, lesiones osteolíticas60–63.
La neuropatía habitualmente se presenta como sensitivo-motora, distal, de curso progresivo y tipo axonal. Se ha descripto también un patrón de neuropatía multifocal, fibras finas dolorosas o síndrome del túnel carpiano debido a infiltración amiloide60.
Neuropatía y gammapatía monoclonalLa neuropatía periférica es una complicación bien conocida de la GM. Esta asociación fue inicialmente identificada en pacientes con MW, en quienes se observaron síntomas de neuropatía asociada en el 8% de los casos64. A la inversa, cerca del 10% de pacientes con neuropatía en estudio sin diagnóstico etiológico presentan GM65. No obstante, dada la alta prevalencia de GM en la población general, la mera presencia de proteínaM en un paciente con neuropatía requiere un profundo análisis de las características de la gammapatía y la neuropatía antes de atribuir causalidad a la misma. En la mayoría de los casos, es necesario descartar otras posibles etiologías de la neuropatía.
Diagnóstico de GMLa electroforesis con inmunofijación es un método de detección más sensible que la electroforesis de proteínas y es el método de elección. El estudio urinario para la detección de proteinuria de Bence-Jones añade sensibilidad al estudio. En caso de detección de proteína monoclonal, ambos tipos de cadenas, pesadas y livianas, deben ser cuantificadas66. En los pacientes con paraproteinemia se debe investigar la presencia de neoplasia hematológica. Estos estudios incluyen, en primera instancia, hemograma completo, frotis y, en algunos casos, biopsia de médula ósea.
Características de los anticuerposSi bien IgG es la proteínaM más común en la población general, cuando se trata de GMSI asociada a neuropatía, la proteínaM más comúnmente hallada es IgM (IgM 60%, IgG 30%, IgA 10%), generalmente asociada a cadenas kappa67.
En la mayoría de los casos se considera que la neuropatía está relacionada con la reactividad del anticuerpo de la proteínaM contra antígenos del nervio. Los determinantes antigénicos identificados incluyen la glucoproteína asociada a la mielina (MAG), el sulfátido, el condroitín-sulfatoC (ChS-C) y algunos gangliósidos68.
- 1.
Anticuerpos anti-MAG
En el 40-50% de los pacientes con GM IgM y neuropatía, la proteínaM se une a MAG.
MAG es una glucoproteína asociada a la mielina del sistema nervioso central y periférico, su peso molecular es de 110.000kDa y se localiza en membranas periaxonales, áreas de mielina no compacta y mesaxones internos y externos. A MAG se le adjudica un rol en la interacción y adhesión mielina-axón69,70.
Inicialmente identificado por Latov et al.71, el anticuerpo anti-MAG reconoce el carbohidrato epítope HNK-1 de MAG. Este componente también está en otros glucoconjugados del nervio periférico, como el sulfátido glucuronil paraglobósido (SGPC) y el sulfátido glucuronil lactosaminil paraglobósido (SGLPC)72.
La detección de estos anticuerpos se realiza por ELISA o Western blot. El método de ELISA es más sensible pero menos específico73. Títulos de 1/3.200 o menos pueden ser identificados en individuos sanos.
- 2.
Anticuerpos antisulfátido
El sulfátido (galactosylceramida-3-0-sulfato) es el mayor glucoesfingolípido de la mielina del sistema nervioso central y periférico y participa en la organización y mantenimiento de la vaina de mielina. Se ha identificado la presencia de títulos elevados de anticuerpos antisulfátidos en pacientes con neuropatía asociada a GM74.
Se trata de la segunda reactividad neural más frecuentemente identificada en pacientes con neuropatía asociada a GM IgM, siendo hallada en entre el 4 y el 8% de los pacientes. Una considerable proporción de los pacientes tiene altos títulos de anti-MAG asociados75.
El método de determinación comúnmente utilizado es por ELISA y los valores de referencia son entre 1/2.000 y 1/16.000. La utilidad de estos anticuerpos es cuestionable dado que rara vez proveen información con implicancias diagnósticas76.
- 3.
Anticuerpos antigangliósidos
Los gangliósidos son glucoesfingolípidos que contienen ácido siálico, particularmente abundantes en las membranas neurales en el sistema nervioso central y periférico.
La relación entre los anticuerpos antigangliósidos y la GM es controvertida dado que la mayoría de los casos de neuropatía asociada a estos anticuerpos ocurren en ausencia de GM.
Se encuentra bien caracterizado el caso de la neuropatía asociada a GM con anticuerpos antigangliósidos que contienen grupos disialosyl (GQ1b, GD1b, GT1b, GD3, GD2)77. Esta reactividad se encuentra en aproximadamente el 2% de los casos de neuropatía asociada a GM IgM y su presencia se asocia a cuadros de neuropatía crónica sensitiva atáxica con compromiso de nervios craneales: CANOMAD (ver presentación clínica). Estos anticuerpos son detectados por ELISA.
FisiopatologíaLa fisiopatología de la neuropatía asociada a GM no está del todo aclarada; se debería a un mecanismo directo de la proteínaM sobre el nervio periférico desencadenando un proceso desmielinizante. Estudios patológicos muestran pérdida de fibras mielínicas y degeneración axonal con depósitos de paraproteína IgM y complemento en la mielina del nervio. La microscopia electrónica muestra separación de las láminas de mielina55,78,79.
Presentación clínicaDADS-M
En general, la neuropatía asociada a paraproteínaM isotipo IgM se presenta como una polineuropatía adquirida, distal y simétrica (DADS-M)60. Típicamente afecta a hombres entre la 6.a y 9.a década de la vida y principalmente compromete fibras gruesas sensitivas causando ataxia y, en ocasiones, temblor. El compromiso motor generalmente está ausente o es de grado leve. El curso suele ser lentamente progresivo con escaso deterioro funcional. La respuesta a la inmunoterapia generalmente es pobre. Desde el punto de vista electrofisiológico, se observan signos compatibles con una polineuropatía desmielinizante con marcado compromiso de las latencias distales, en forma desproporcionada a la caída de la velocidad de conducción en los segmentos proximales del nervio; los bloqueos de conducción son excepcionales. En los pacientes que presentan este fenotipo, los anticuerpos anti-MAG se identifican en el 50% de los casos, sin embargo, no se hallan diferencias en el tipo ni en la severidad de la neuropatía respecto de los pacientes con DADS-M pero anti-MAG negativos61.
Gammapatía monoclonal (GM) no-IgM
En contraste, los pacientes con GM no-IgM se presentan con un espectro más amplio de fenotipos: desde neuropatía axonal sensitivo-motora hasta PIDC. La PIDC-GMSI tiene las mismas características clínicas y electrodiagnósticas que la PIDC pura y responde de igual forma al tratamiento.
CANOMAD
Otro fenotipo, menos frecuente, asociado a IgM-GMSI es el CANOMAD (neuropatía atáxica crónica con oftalmoplejía, proteínaM, aglutininas frías y anticuerpos antidisialosyl). Esta neuropatía crónica se asocia con compromiso de nervios periféricos y craneales. Se presenta generalmente en la 5.a década y es más frecuente en hombres. El curso es generalmente crónico progresivo, pero algunos pacientes presentan un patrón en recaídas y remisión. El diagnóstico se basa en el cuadro clínico y en la presencia de paraproteína IgM que reaccionan contra gangliósidos disialosilados: GD1b, GD3, GQ1b y GT1b. Cerca del 50% de estos anticuerpos IgM son aglutininas frías77.
Otras enfermedades asociadas a paraproteínas: POEMS y amiloidosis AL.
POEMS
El síndrome POEMS es un desorden clonal de células plasmáticas denominado por el acrónimo de sus manifestaciones (polineuropatía - organomegalia - endocrinopatía - proteína monoclonal - cambios dérmicos). Esta entidad se asocia a mieloma osteoesclerótico y niveles levemente aumentados de inmunoglobulinas IgA o IgG-lambda en el 90% de los pacientes. La neuropatía es el elemento principal de la enfermedad y a menudo precede el diagnóstico del mieloma; su curso es crónico progresivo con compromiso sensitivo y motor, con patrón desmielinizante, pero con más severo compromiso axonal sobreagregado (reducción en amplitudes motoras, presencia de fibrilaciones) que lo observado en DADS-M o PIDC. Una evaluación ósea extensa radiográfica o tomográfica permitirá identificar el foco osteoesclerótico54. Otras características incluyen organomegalia (esplenomegalia o hepatomegalia), hiperpigmentación y edemas. Los niveles de VEGF (factor de crecimiento endotelial vascular) se encuentran marcadamente elevados y se correlacionan con la actividad de la enfermedad. El mecanismo subyacente a la neuropatía es desconocido. Algunas hipótesis postulan aumento de la permeabilidad microvascular y edema endoneural, daño endotelial y microangiopatía secundaria o activación de la cascada del complemento por proteínaM generando disrupción de la barrera hemato-nerviosa60–66.
Amiloidosis primaria
Es un desorden multisistémico en el cual fibras amiloides secundarias a la presencia de cadenas livianas libres se depositan en varios órganos, incluyendo riñones, hígado, tracto gastrointestinal y nervios periféricos. Entre el 15-20% de pacientes con amiloidosis primaria cursan con neuropatía periférica progresiva, distal, dolorosa y axonal, con compromiso de fibras finas, que comienza en extremidades inferiores y asocia compromiso autonómico. Neuropatías por atrapamiento, como síndrome del túnel carpiano, son observadas con frecuencia. A diferencia de la neuropatía asociada a GM, que puede mantenerse estable por temporadas, la neuropatía amiloidea tiene un curso más progresivo y discapacitante. El diagnóstico definitivo requiere la demostración de los depósitos amiloides en la biopsia de nervio, visualizados con tinción de rojo Congo. El posible mecanismo en esta neuropatía es secundario a la insuficiencia vascular debido al depósito de amiloide en los vasos sanguíneos o a un efecto tóxico directo sobre las fibras nerviosas60,61.
Anticuerpos en ganglionopatías y neuronopatíasVarios términos se utilizan para referirse a las ganglionopatías y neuronopatías. Algunos autores proponen unificar la nomenclatura por motivos anatómicos y clínicos80.
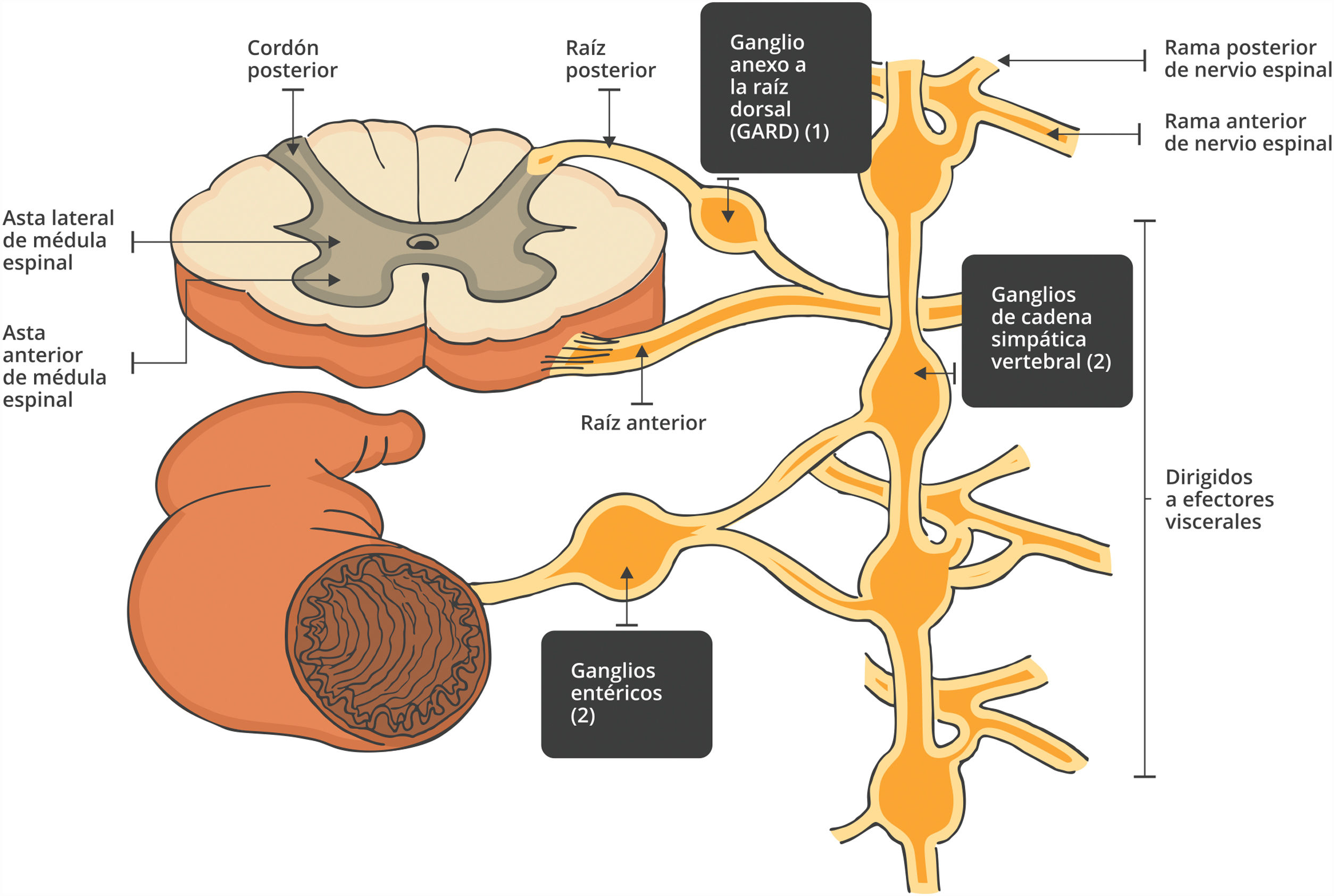
Anatómicamente, se diferencian los ganglios anexos a la raíz dorsal (GARD) de los ganglios pertenecientes al sistema nervioso autónomo. El GARD es la estación de relevo de las vías sensoriales (fig. 4). Las proyecciones periféricas de las neuronas del GARD serán las fibras sensitivas de los nervios periféricos81–83. Los ganglios autonómicos (simpáticos, parasimpáticos y entéricos) se encuentran en la región paravertebral o cerca de los órganos que inervan, y dan origen a fibras posganglionares que inervan el miocardio, el músculo liso y las glándulas84–86.
 anti-Hu, anti-CV2/CRMP-5, ANNA y anti-SSA/SSB; en (2): anti-AChR ganglionares.")
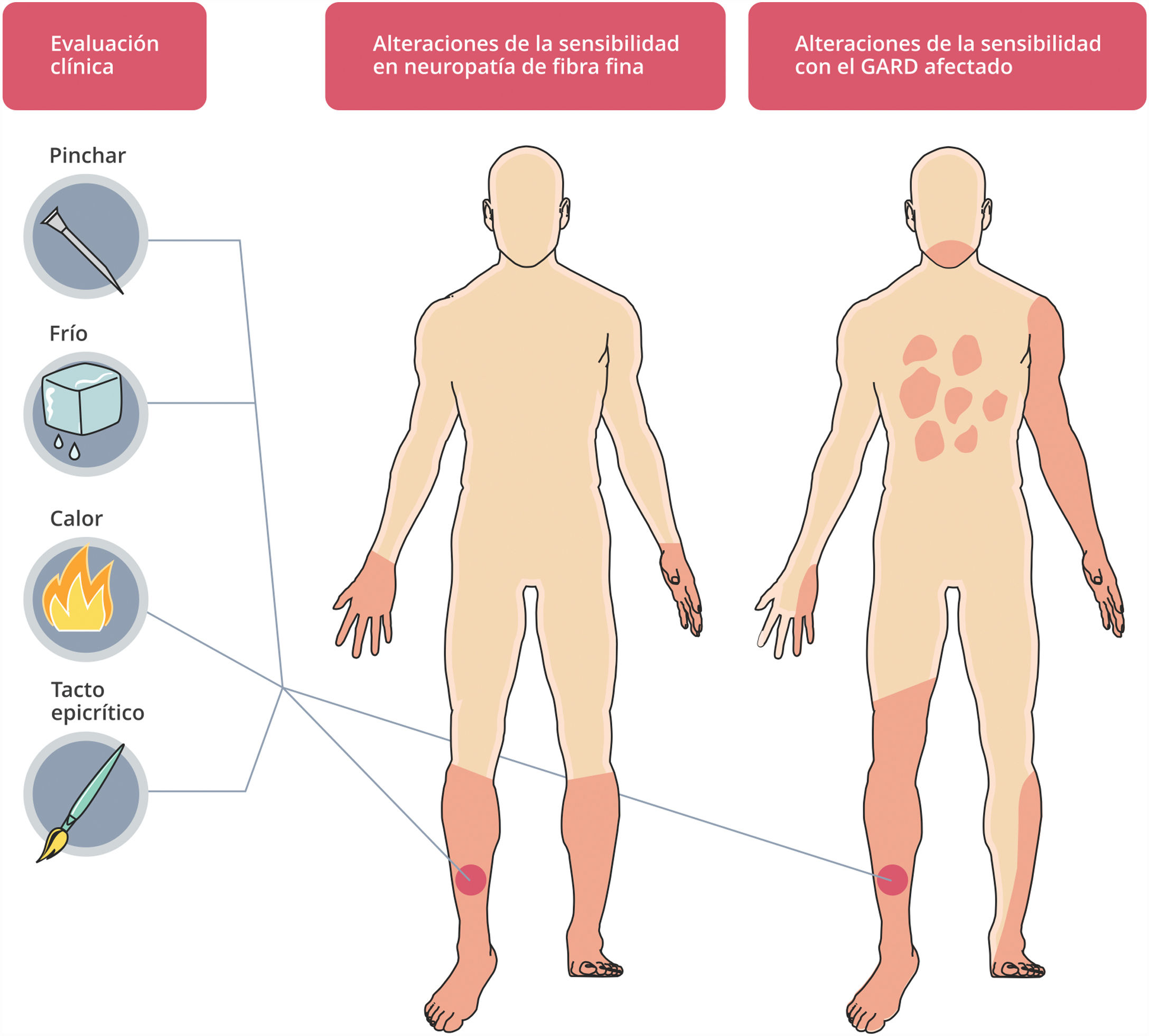
Clínicamente las neuropatías más frecuentes provocan compromiso bilateral y simétrico de nervios periféricos, que progresan de distal a proximal. Esto contrasta con el patrón asimétrico observado cuando hay compromiso del GARD (fig. 5).
.")
Utilizaremos el término «neuronopatía sensitiva» para describir el compromiso del GARD; «ganglionopatía autonómica», para el compromiso de los ganglios del sistema nervioso autónomo, y por último «polineuropatía sensitiva» cuando hay compromiso clínico distal y simétrico.
Neuronopatías sensitivas (asociadas al GARD)Denny-Brown describió esta entidad en 2 pacientes con carcinoma bronquial y severa lesión de neuronas del GARD87. Lo llamó neuropatía sensorial primaria y detalló el cuadro clínico de ataxia sensorial. Otros términos se han utilizado para esta entidad: neuronopatía sensitiva, ganglionopatías, poliganglionopatías, enfermedad de la neurona sensitiva y neuropatía sensitiva-atáxica80,88,89.
Existen diferentes causas: paraneoplásicas, infecciosas, tóxicas, idiopáticas y autoinmunes como el síndrome de Sjögren, lupus eritematoso sistémico, hepatitis autoinmune y enfermedad celíaca89 (tabla 2).
Causas de neuronopatías adquiridas (asociadas al GARD)
| Comienzo | Causa | Anticuerpos asociados | |
|---|---|---|---|
| Paraneoplásica | Subagudo Crónico | Cáncer de pulmón de células pequeñas, carcinoma bronquial, cáncer de ovario, cáncer de próstata, tumor neuroendocrino, sarcoma, carcinoma hepatocelular | Anti-Hu, anti-CRMP-5/CV2 |
| Autoinmune | Subagudo Crónico | Síndrome de Sjögren, LES, enfermedad celíaca, hepatitis autoinmune, neuronopatía asociada a anticuerpo anti-FGFR3 | ANNA, anti-SSA, anti-SSB, anti-dsDNA, anti-TTG, antigliadina, anti-FGFR3 |
| Infecciosa | Subagudo | HIV, EBV, VZV, HTLV1, lepra, enterovirus | |
| Tóxica | Subagudo Crónico | Piridoxina, platinos | Estudios de laboratorio, vitamina B6, historia de exposición a platinos |
| Idiopática | Crónico | Autoinmune? | Diagnóstico de exclusión |
ANNA: anticuerpo antinuclear; CRMP-5: proteína mediadora de la respuesta a la colapsina; dsDNA: DNA de doble cadena; EBV: virus Epstein-Barr; FGFR3: factor de crecimiento fibroblástico 3; GARD: ganglio anexo raíz dorsal; HIV: virus de la inmunodeficiencia humana; HTLV: virus linfotrófico T humano; LES: lupus eritematoso sistémico; TTG: transglutaminasa tisular; VZV: virus varicela zoster.
Clínicamente se caracteriza por pérdida sensitiva progresiva no restringida a miembros inferiores, a menudo indolora, suelen tener ataxia sensorial grave y temprana. Los síntomas generalmente son multifocales y asimétricos. Si la pérdida propioceptiva es severa puede haber pseudoatetosis. Los estudios electrofisiológicos muestran respuestas sensitivas reducidas o ausentes con respuestas motoras preservadas.
Los anticuerpos que con mayor frecuencia están involucrados son: anti-Hu (ANNA-1); anti-CV2/CRMP-5; ANNA; anti-SSA/SSB (anti-Ro y anti-La, respectivamente).
Anticuerpos anti-Hu (anticuerpo ANNA-1)Su antígeno, ELAVL (Hu), se localiza en los núcleos y el citoplasma de neuronas del sistema nervioso central y periférico y células tumorales. Son anticuerpos contra una familia de proteínas de unión al ARN (Hu-D, Hu-C y Hel N1) las cuales tienen una importante función en el desarrollo y mantenimiento de las neuronas. No son patogénicos, pero en pacientes con neuronopatía la identificación de los mismos obliga a buscar cáncer de pulmón de células pequeñas (CPCP)90,91. También se asocian a neuropatía sensitiva, encefalomielitis y degeneración cerebelosa con disfunción autonómica y con encefalitis límbica. Tienen una sensibilidad del 80% para identificar CPCP. Otros tumores asociados son carcinoma de próstata, neuroblastoma, cáncer de mama y sarcoma. Los pacientes con anticuerpos anti-Hu son en su mayoría varones con un mal pronóstico y con una severa e irreversible discapacidad neurológica. Por IFI se marca el núcleo de las neuronas del sistema nervioso central y periférico. Se realiza la confirmación mediante inmunotransferencia92.
Existe una entidad llamada «neuropatía entérica paraneoplásica». Es un subconjunto de pacientes anti-Hu positivos con compromiso severo de neuronas entéricas. La característica clínica es la gastroparesia y puede coexistir con neuronopatía sensorial.
Anticuerpos anti-CV2/CRMP-5El antígeno diana es una proteína citosólica denominada «proteína mediadora de la respuesta a colapsina» que se corresponde con la CV2 (CRMP-5). Este anticuerpo presenta algunas características comunes con los ANNA-1 en cuanto al cuadro clínico y la neoplasia asociada. Como otros anticuerpos dirigidos a antígenos citoplasmáticos, su rol fisiopatológico es discutible, y podrían ser solo marcadores de la respuesta inmune citotóxica de células T hacia las neuronas. Los anti-CV2 están vinculados a una amplia gama de síndromes neurológicos (encefalomielitis, degeneración cerebelosa, corea, neuronopatía y neuropatía sensitivo-motora). Se asocian a CPCP y a timoma subyacentes en aproximadamente el 80% de los casos93. La supervivencia de los pacientes es considerablemente más larga (48 meses) en comparación con los ANNA-1 (11 meses). Si hay coexistencia de anti-CV2 (CRMP-5) y ANNA-1 en un mismo paciente, la supervivencia media es alrededor de 18 meses. Por IFI se tiñe el citoplasma de oligodendrocitos de la capa granular y la sustancia blanca del cerebelo. Sin embargo, es relativamente difícil de detectar.
Anticuerpos antinucleares (ANNA)Los ANNA tienen como blanco el contenido nuclear. Si bien se encuentran en bajas concentraciones en la población general, existe un 5% que posee títulos elevados (especialmente mujeres); la mitad de estos desarrolla una patología autoinmune. Los ANNA se pueden asociar a neuronopatía sensitiva91. Se detectan por IFI utilizando células de la línea HEp-2 y tienen patrones de tinción asociados a diferentes enfermedades94.
Con títulos altos de ANNA, se procede, según el patrón obtenido, a determinar los subtipos implicados, como por ejemplo los anti-ENA (antígenos nucleares extraíbles), y dentro de estos, los anti-Ro (SS-A) y anti-La (SS-B). Para confirmar la especificidad de estos anticuerpos se utiliza inmunotransferencia. Si la sospecha clínica de enfermedad autoinmune lo ameritara, se solicitarían estos últimos anticuerpos aun con ANNA negativos94.
Ganglionopatía autonómica autoinmune (AAG) o insuficiencia autonómica autoinmuneEn los ganglios autonómicos se encuentran receptores colinérgicos diferentes de los que se encuentran en el músculo esquelético. Estos receptores, nicotínicos, median la transmisión sináptica rápida en los ganglios simpáticos, parasimpáticos y entéricos.
El receptor de acetilcolina (AChR) es una proteína compuesta por 5 subunidades, 2 de ellas denominadas α, y las restantes β, γ y δ.
Los anticuerpos IgG específicos para el AChR nicotínico ganglionar (G-AChR) se encuentran en el 50% de los pacientes con falla panautonómica pura aguda o subaguda; la seropositividad para estos confirma el diagnóstico de ganglionopatía autonómica autoinmune (AAG) y muchas veces permite identificar neoplasias e instaurar el tratamiento inmunomodulador95–98.
Los anticuerpos contra G-AChR inhiben específicamente las corrientes de membrana a través de la subunidad α3 del AChR. Vale destacar que estos anticuerpos son diferentes al anticuerpo contra AChR que se solicita en los casos de miastenia grave, el reactivo específico para G-AChR ganglionar no se encuentra aún disponible en nuestro país. La reactividad cruzada de anticuerpos contra los diferentes AChR es poco común, pero puede suceder99,100.
La clínica de AAG es una pan-disautonomía grave que incluye: pupilas fijas, anhidrosis, ortostatismo severo, inmovilidad gastrointestinal severa, disfunción sexual y alteraciones urinarias. Los casos típicos de AAG tienen una presentación monofásica inicial, y pueden experimentar mejoría espontánea parcial.
Existen otras patologías asociadas a anticuerpos contra AChR ganglionar. El riesgo de adenocarcinoma de timo es del 10 al 30% (especialmente en superposición con miastenia grave)97.
El hallazgo de anticuerpos anti-AChR ganglionares (subunidades α3 β4) confirma el diagnóstico de AAG, y los títulos correlacionan con la gravedad clínica99,100.
Polineuropatías sensitivas (neuropatías de fibras finas)Las neuropatías de fibra fina se caracterizan por el compromiso de fibras aferentes Aδ y C amielínicas. Tienen una incidencia de 12/100.000 habitantes por año y una prevalencia de 53/100.000 habitantes101–104.
Se caracterizan por disfunción autonómica y polineuropatía con distribución distal, bilateral y simétrica de las extremidades y fenómenos positivos o negativos104,105.
Las causas son: metabólicas, deficiencia o intoxicación vitamínica, tóxicos, infecciones, hereditarias e inmunológicas (enfermedad celíaca, GM, amiloidosis primaria, síndrome paraneoplásico, sarcoidosis, esclerodermia, síndrome de Sjögren, lupus eritematoso sistémico y vasculitis). No hay un anticuerpo específico.
Conflicto de interesesEl grupo de trabajo no presenta conflictos de interés con relación a la redacción de este artículo.
Mariana Bendersky. Servicio de Neurología Infantil, Hospital Italiano de Buenos Aires, Instituto Argentino de Investigaciones Neurológicas (IADIN)-Facultad de Medicina, Universidad de Buenos Aires-EnyS-CONICET.
