



La enfermedad de Creutzfeldt-Jakob esporádica (ECJe) es la forma de presentación más frecuente de las enfermedades producidas por priones. Las características clínicas de esta entidad están ampliamente descritas1,2, no obstante, la presencia de afectación de segunda motoneurona es una forma de manifestación rara, y aún más si esta se presenta al inicio de la enfermedad3.
Varón de 54 años previamente sano, que refiere historia de 4 semanas de evolución, con torpeza e inestabilidad para caminar. En la exploración neurológica se objetivó fasciculaciones en deltoides, bíceps, tríceps y cuádriceps, con amiotrofia y debilidad leve de musculatura proximal de miembros, principalmente cuádriceps, deltoides y musculatura periescapular. También se objetivó arreflexia global con leve dismetría en los 4 miembros de predominio izquierdo, temblor ortostático y marcha atáxica con tandem imposible. No habían signos de afectación de primera motoneurona.
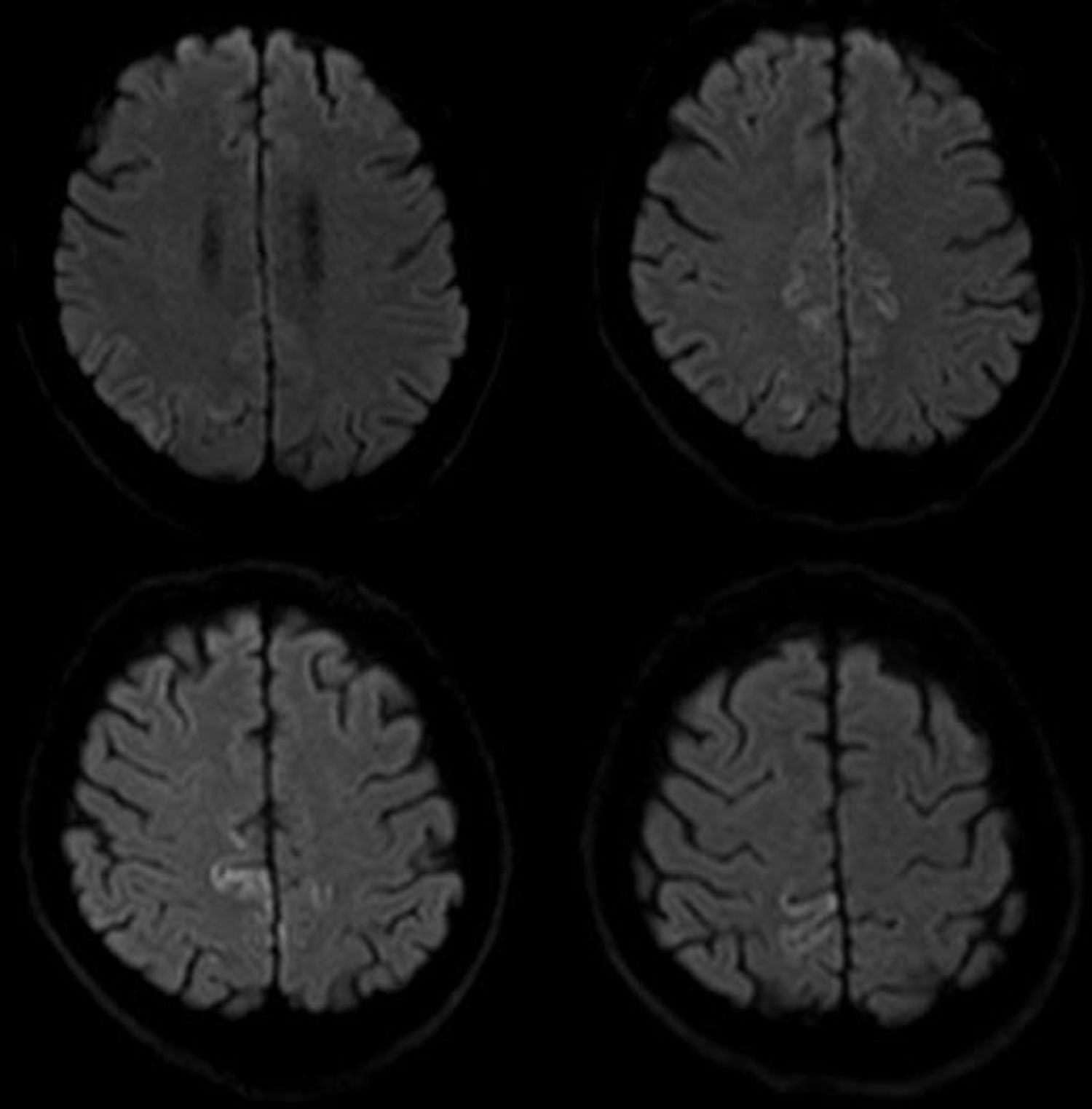
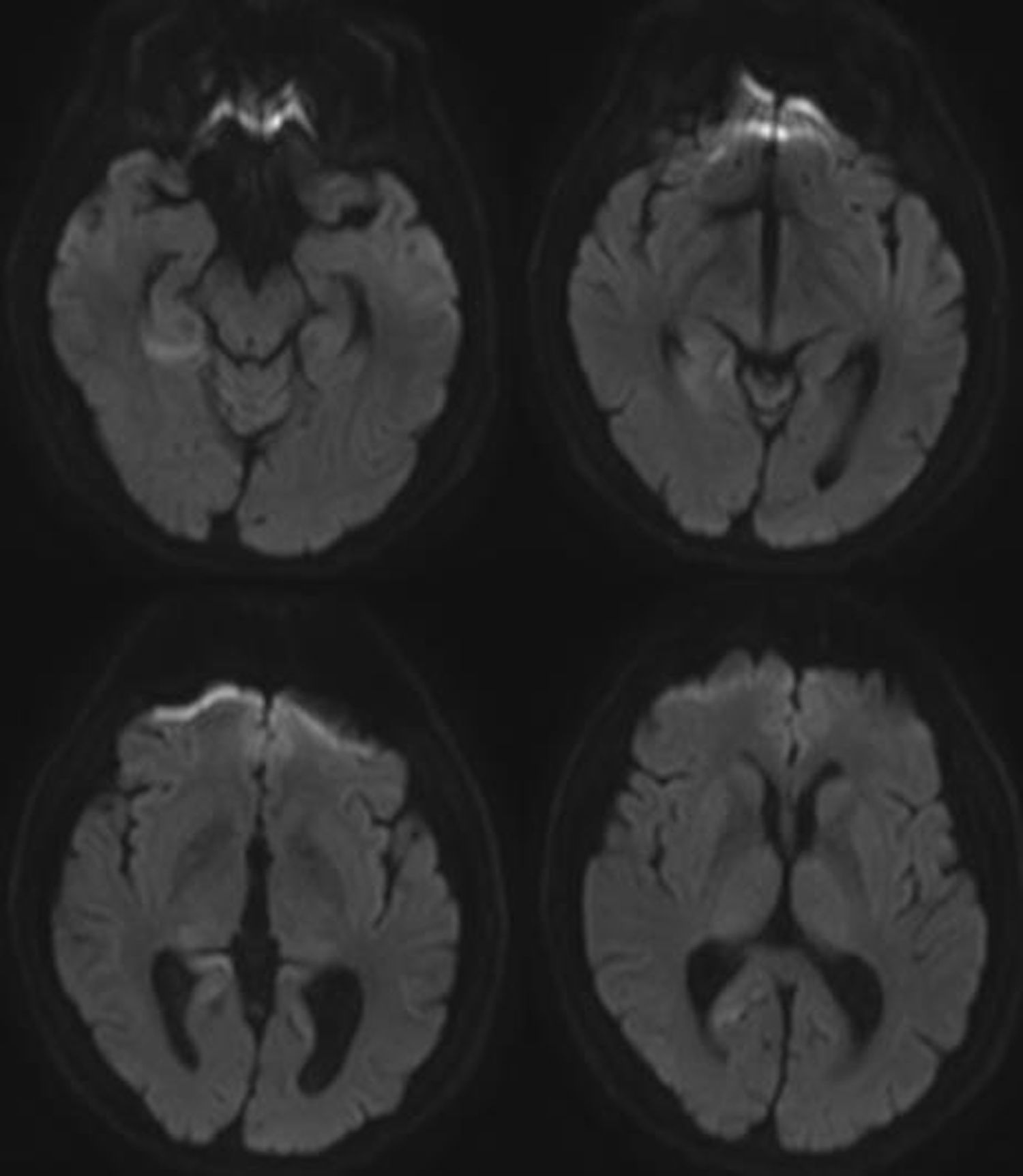
Las pruebas de imagen incluyeron RMN cerebral que reveló alteraciones corticales únicamente en secuencias de difusión (fig. 1). Las pruebas de laboratorio abarcaron hemograma, bioquímica, función renal, hepática y tiroidea, vitamina B12 y ácido fólico que fueron normales. También se incluyó estudio infeccioso, anticuerpos paraneoplásicos, marcadores tumorales y autoinmunidad que resultaron negativos. Se realizó un electroencefalograma que únicamente mostró leve desorganización difusa del trazado y un electromiograma que denotó amplia afectación denervativa en forma de fibrilaciones, ondas positivas y fasciculaciones en todos los músculos explorados, con estudio de conducción nerviosa normal. El estudio neuropsicológico objetivó alteraciones visuoespaciales leves, déficit de la memoria reciente y reducción de la fluencia verbal congruentes con deterioro cognitivo de perfil córtico-subcortical. El análisis del líquido cefalorraquídeo mostró positividad para la proteína 14-3-3 y elevación de proteína TAU. Una RMN cerebral, un mes después, objetivó nuevas áreas de alteración de señal a nivel cortical en secuencias de difusión (fig. 2). Clínicamente el paciente evolucionó hacia un deterioro cognitivo severo y una ataxia invalidante; además, las fasciculaciones se hicieron visibles en musculatura distal de extremidades, sin afectación facial ni lingual. En última instancia, el paciente desarrolló mutismo acinético y falleció, aproximadamente, 15 meses después del diagnóstico.


La ECJe se caracteriza clínicamente por el desarrollo de demencia rápidamente progresiva, ataxia, alteración del tono muscular y mioclonías3, no obstante, pueden no estar presentes todas estas características y manifestarse de forma atípica. Así pues, como en este paciente, se ha descrito de forma aislada la presencia de amiotrofia, fasciculaciones y arreflexia como parte del espectro clínico de esta enfermedad4,5, no obstante es excepcional su presentación al inicio de la clínica6,7. El mecanismo fisiopatológico de esta rara forma de expresión es difícil de dilucidar dado que los estudios postmortem únicamente suelen incluir el cerebro, sin embargo, se ha sugerido una pérdida de células del asta anterior medular secundaria a degeneración espongiforme como mecanismo causal4,7,8. Clásicamente se ha definido la variante clínica amiotrófica9 y la presencia de afectación de segunda motoneurona se ha descrito en las formas familiares y esporádicas de la enfermedad; no obstante, esta no se ha asociado a ninguna forma genética en concreto y el único caso donde se realizó análisis genotípico mostró la variante 129M/M6.
En cuanto a las pruebas de imagen, señalar que la presencia de hiperintensidad cortical aislada puede hallarse hasta en un tercio de los pacientes con diagnóstico final de ECJe10. En este sentido, acorde con nuestro caso, la evidencia disponible sugiere que la secuencia de difusión es la más sensible para la detección de cambios espongiformes cerebrales producidos en esta prionopatía, particularmente en las fases más tempranas de la enfermedad11–13.
Así, aunque la presencia de afectación de motoneurona nos hizo ampliar el diagnóstico diferencial, la presencia de afectación cerebelosa, el deterioro cognitivo incipiente, y sobre todo los hallazgos de neuroimagen nos permitieron un diagnóstico etiológico temprano. Asimismo, la ausencia de signos de neurona motora superior, la falta de clínica bulbar y la rápida progresión hacen poco probable la coexistencia de esclerosis lateral amiotrófica.
En conclusión, aunque la presencia de afectación de segunda motoneurona es una manifestación rara en los pacientes con ECJe, es necesario no solo considerarla dentro del espectro clínico de esta enfermedad sino tener en cuenta que esta puede ser una manifestación temprana que nos puede ayudar a alcanzar un diagnóstico precoz.
FinanciaciónNo se ha recibido financiación para la realización del trabajo.

