





Amyotrophic lateral sclerosis (ALS) is an insidious, clinically heterogeneous neurodegenerative disease associated with a diagnostic delay of approximately 12 months. No study conducted to date has analysed the diagnostic pathway in Spain.
MethodsWe gathered data on variables related to the diagnostic pathway and delay for patients diagnosed with ALS between October 2013 and July 2017.
ResultsThe study included 143 patients with ALS (57% men; 68% spinal onset). Patients were diagnosed in public centres in 86% of cases and in private centres in 14%. The mean diagnostic delay was 13.1 months (median 11.7). Patients were examined by neurologists a mean time of 7.9 months after symptom onset, with diagnosis being made 5.2 months later. Half of all patients underwent unnecessary diagnostic tests and multiple electrophysiological studies before diagnosis was established. Diagnostic delay was longer in cases of spinal onset (P=.008) due to onset of the disease in the lower limbs. No differences were found between the public and private healthcare systems (P=.897).
ConclusionsThe diagnostic delay in ALS in Spain is similar to that of neighbouring countries and seems to depend on disease-related factors, not on the healthcare system. Patients with lower-limb onset ALS constitute the greatest diagnostic challenge. Misdiagnosis is frequent, and partly attributable to an incorrect approach or erroneous interpretation of electrophysiological studies. Specific training programmes for neurologists and general neurophysiologists and early referral to reference centres may help to reduce diagnostic delay.
La esclerosis lateral amiotrófica (ELA) es una enfermedad insidiosa y clínicamente heterogénea, lo que resulta en un retraso diagnóstico de unos 12 meses. En España el trayecto diagnóstico no ha sido analizado.
MétodosSe recogieron variables relativas al trayecto y retraso diagnóstico de pacientes diagnosticados de ELA entre octubre del 2013 y julio del 2017.
ResultadosSe incluyó a 143 pacientes con ELA (57% varones, 68% de inicio espinal). El 86% de ellos fueron estudiados en centros públicos y un 14% en privados. El retraso diagnóstico medio fue de 13.1 meses (mediana 11.7). El paciente tardó de media 7.9 meses en llegar al neurólogo y este, 5,2 meses más en diagnosticarlo. En la mitad de los pacientes se realizaron pruebas innecesarias y más de un estudio electrofisiológico para llegar al diagnóstico. El retraso diagnóstico fue mayor en los casos espinales (p=0,008), atribuible a los pacientes cuyos síntomas se iniciaron en miembros inferiores, pero sin diferencias entre el sistema público y privado (p=0,897).
ConclusionesEl retraso diagnóstico de la ELA en nuestro medio es similar al de países de nuestro entorno y parece determinado por factores propios de la enfermedad e independiente del sistema sanitario. Las formas de inicio en miembros inferiores constituyen el mayor reto. Los errores diagnósticos del neurólogo son frecuentes y en parte atribuibles a una mala orientación o interpretación del estudio electrofisiológico. La formación específica del neurólogo y neurofisiólogo general y la derivación precoz a centros de referencia podrían ayudar a reducir la demora.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterised by progressive weakness secondary to spinal and cortical motor neuron death, with patients typically dying due to respiratory insufficiency within 3-4 years of symptom onset.1
Although the disease progresses rapidly, early diagnosis can be highly challenging due to its insidious onset and clinical heterogeneity, and the lack of diagnostic markers.2 This frequently results in misdiagnosis, unnecessary testing, and a diagnostic delay of approximately a year,3–13 or roughly one-third of the overall survival time. This diagnostic delay appears to be independent of country and health system,2–10,14 and has not significantly changed in the last 20 years despite various strategies11 and the proposed revisions to the El Escorial diagnostic criteria.2,15
For a disease as devastating as ALS, reducing diagnostic delay may positively impact patients and their relatives, reducing uncertainty and anxiety during the diagnostic process. Early diagnosis limits the number of unnecessary tests and interventions performed, enables early treatment with riluzole (when this approach is most likely to be effective), improves the ability to plan for the future, and facilitates patients’ inclusion in clinical trials at earlier stages of the disease.2
To date, no Spanish study has analysed the diagnostic pathway of patients with ALS. In this article, we describe the diagnostic pathway and diagnostic delay in patients with ALS in our setting and compare them against results from other countries.
Patients and methodsPatientsThis study included consecutive patients attended at the ALS unit at Hospital La Fe (Valencia, Spain) and diagnosed with ALS between October 2013 and July 2017.
Hospital La Fe is a tertiary hospital that receives patients referred from other public and private centres for diagnosis or second opinions, as well as correctly diagnosed patients who wish to be treated at the hospital's multidisciplinary ALS unit. Therefore, some patients included in the study were diagnosed at Hospital La Fe, while others were diagnosed elsewhere.
For the purposes of the study, we selected only those patients with a clinical diagnosis of ALS; patients not found to meet the El Escorial criteria at the first assessment were followed up for at least 6 months and were excluded from the study if the diagnosis could not be confirmed. We also excluded patients who were under neurological follow-up for cognitive impairment prior to the onset of motor symptoms, as well as those diagnosed with progressive muscular atrophy or primary lateral sclerosis (defined as at least 4 years’ history of alterations exclusively affecting the upper or the lower motor neurons, respectively).16 Finally, we excluded patients for whom insufficient data were available on the diagnostic process.
Study variablesWe performed a descriptive, cross-sectional study based on data collected both prospectively and retrospectively. At the first consultation, one researcher (JFVC) prospectively collected demographic and clinical data: age, sex, referring hospital, date and region of symptom onset, progression rate,17 and Awaji category at diagnosis.15 Three researchers (JFVC, MMM, and MFP) collected retrospective data on aspects of the diagnostic pathway by reviewing medical notes from each consultation on the hospital's IT system (the Orion Clinic program) and the Valencian Healthcare Agency's outpatient care information system (SIA-Gaia), as well as records from hospitals not using these systems, which were provided by the patients. The variables studied included: first physician consulted and date of initial consultation; specialist consultations and dates; tests performed (classified as necessary and unnecessary, according to the European diagnostic guidelines2); the dates of electrophysiological (EP) studies; emergency department visits; diagnosis established by the neurology department; date of diagnosis; and date of riluzole treatment onset. Supplementary Tables 1 and 2 (Appendix 1) show the data for each of the retrospective and prospective variables studied. Doubts arising during retrospective data collection were resolved by consensus between the 3 researchers. For patients who reported consulting a physician within the first 15 days after symptom onset, we reviewed their medical histories in detail to identify the symptoms that led them to seek medical attention so rapidly. Finally, JFVC reviewed all the retrospective data.
Statistical analysisStatistical analysis was conducted using the R and RStudio software (version 3.3.1). For the descriptive analysis, we calculated means and medians (with standard deviations and first and third quartiles, respectively) for quantitative variables and percentages for qualitative variables. For the inferential analysis, we used a linear regression model for continuous quantitative variables (diagnostic delay) and a logistic regression model for discrete variables following a Poisson distribution (number of regions affected in the EP study, according to the Awaji criteria).
Bioethics committeeThe study was approved by Hospital La Fe's clinical research ethics committee. All patients gave written informed consent to the storage and use of their personal data.
ResultsPatient characteristicsOf a total of 186 patients assessed and diagnosed at the hospital's motor neuron disease unit between October 2013 and July 2017, the diagnosis of ALS was confirmed in 159 patients; 143 were included in the final sample after application of the inclusion and exclusion criteria described above (Fig. 1).

Patient selection process. Of a total of 186 patients assessed at the motor neuron disease unit, clinical diagnosis of ALS was confirmed after 6 months’ follow-up in 159. We excluded 27 patients who were diagnosed with other conditions. A further 16 patients were excluded for various reasons, resulting in a final sample of 143. LMN: lower motor neuron; UMN: upper motor neuron.
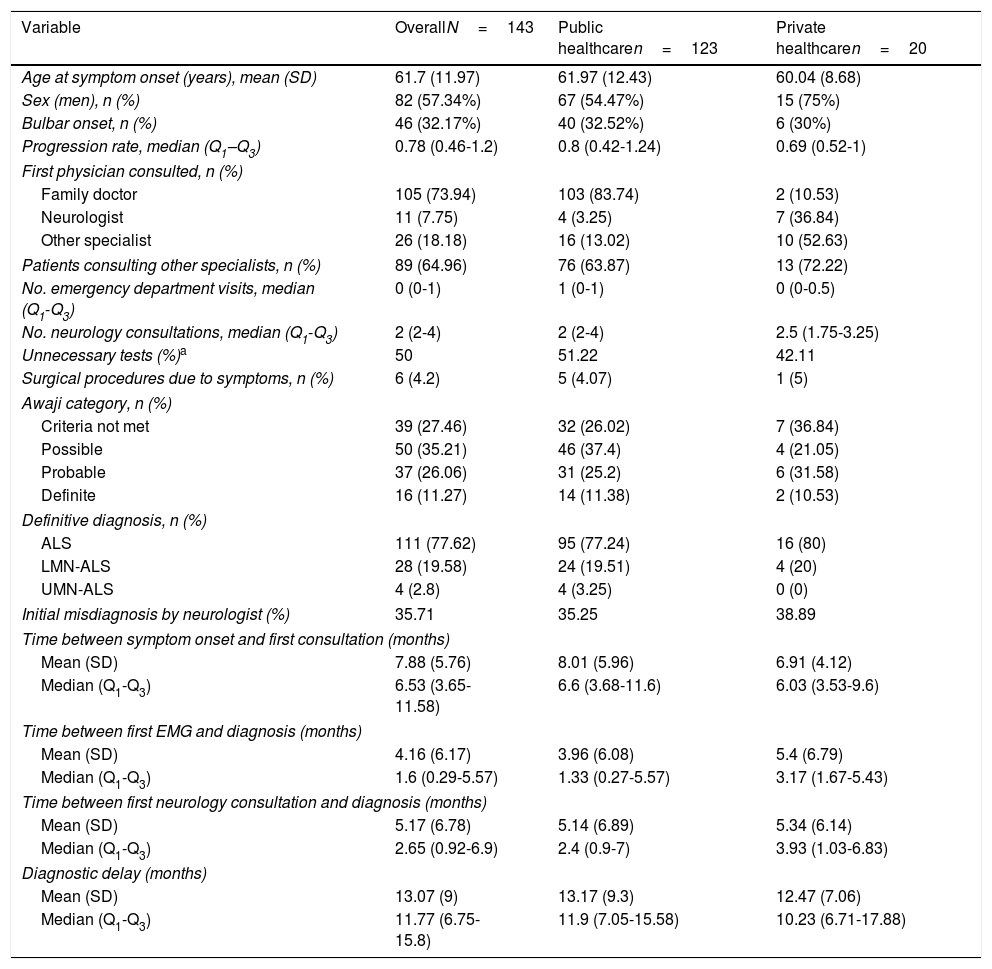
Table 1 shows the demographic, clinical, and treatment pathway data collected. Patients had a mean age of 62 years, were predominantly men, and mainly presented spinal involvement (most frequently affecting the lower or upper limbs [40% and 26%, respectively] and rarely axial or respiratory muscles [1.4% and 0.7%]). Eight percent of patients had family history of ALS.
Patient and diagnostic pathway characteristics.
| Variable | OverallN=143 | Public healthcaren=123 | Private healthcaren=20 |
|---|---|---|---|
| Age at symptom onset (years), mean (SD) | 61.7 (11.97) | 61.97 (12.43) | 60.04 (8.68) |
| Sex (men), n (%) | 82 (57.34%) | 67 (54.47%) | 15 (75%) |
| Bulbar onset, n (%) | 46 (32.17%) | 40 (32.52%) | 6 (30%) |
| Progression rate, median (Q1–Q3) | 0.78 (0.46-1.2) | 0.8 (0.42-1.24) | 0.69 (0.52-1) |
| First physician consulted, n (%) | |||
| Family doctor | 105 (73.94) | 103 (83.74) | 2 (10.53) |
| Neurologist | 11 (7.75) | 4 (3.25) | 7 (36.84) |
| Other specialist | 26 (18.18) | 16 (13.02) | 10 (52.63) |
| Patients consulting other specialists, n (%) | 89 (64.96) | 76 (63.87) | 13 (72.22) |
| No. emergency department visits, median (Q1-Q3) | 0 (0-1) | 1 (0-1) | 0 (0-0.5) |
| No. neurology consultations, median (Q1-Q3) | 2 (2-4) | 2 (2-4) | 2.5 (1.75-3.25) |
| Unnecessary tests (%)a | 50 | 51.22 | 42.11 |
| Surgical procedures due to symptoms, n (%) | 6 (4.2) | 5 (4.07) | 1 (5) |
| Awaji category, n (%) | |||
| Criteria not met | 39 (27.46) | 32 (26.02) | 7 (36.84) |
| Possible | 50 (35.21) | 46 (37.4) | 4 (21.05) |
| Probable | 37 (26.06) | 31 (25.2) | 6 (31.58) |
| Definite | 16 (11.27) | 14 (11.38) | 2 (10.53) |
| Definitive diagnosis, n (%) | |||
| ALS | 111 (77.62) | 95 (77.24) | 16 (80) |
| LMN-ALS | 28 (19.58) | 24 (19.51) | 4 (20) |
| UMN-ALS | 4 (2.8) | 4 (3.25) | 0 (0) |
| Initial misdiagnosis by neurologist (%) | 35.71 | 35.25 | 38.89 |
| Time between symptom onset and first consultation (months) | |||
| Mean (SD) | 7.88 (5.76) | 8.01 (5.96) | 6.91 (4.12) |
| Median (Q1-Q3) | 6.53 (3.65-11.58) | 6.6 (3.68-11.6) | 6.03 (3.53-9.6) |
| Time between first EMG and diagnosis (months) | |||
| Mean (SD) | 4.16 (6.17) | 3.96 (6.08) | 5.4 (6.79) |
| Median (Q1-Q3) | 1.6 (0.29-5.57) | 1.33 (0.27-5.57) | 3.17 (1.67-5.43) |
| Time between first neurology consultation and diagnosis (months) | |||
| Mean (SD) | 5.17 (6.78) | 5.14 (6.89) | 5.34 (6.14) |
| Median (Q1-Q3) | 2.65 (0.92-6.9) | 2.4 (0.9-7) | 3.93 (1.03-6.83) |
| Diagnostic delay (months) | |||
| Mean (SD) | 13.07 (9) | 13.17 (9.3) | 12.47 (7.06) |
| Median (Q1-Q3) | 11.77 (6.75-15.8) | 11.9 (7.05-15.58) | 10.23 (6.71-17.88) |
ALS: amyotrophic lateral sclerosis; LMN: lower motor neuron; Q: quartile; UMN: upper motor neuron.
“Unnecessary tests” were defined as all those tests not indicated for the diagnosis of ALS in the European guidelines2 (e.g., hand radiography) and all tests that were unnecessarily repeated. Tests were categorised by consensus on a case-by-case basis.
Median time between symptom onset and first consultation was 2.5 months; curiously, 20 patients (14%) consulted early, in the first 15 days after onset. Medical records did not specify the initial form of presentation for 7 of these patients; onset was acute in the remaining 13, with 3 different forms of presentation:
- –
Painful onset (8 patients): 6 with lower-limb onset and one with upper-limb onset consulted due to low back pain (4), limb pain (2), or muscle cramps (2). One patient with bulbar onset consulted due to pharyngeal pain.
- –
Postsurgical onset (4 patients): these patients reported symptom onset following surgery (trauma surgery in 3 cases and appendectomy in one). In this group, 3 patients presented lower-limb onset and one presented bulbar onset.
- –
Stroke mimic: one patient presented apparently sudden dysarthria and was referred to our centre by the emergency department physician, who suspected stroke.
A total of 123 patients (86%) were attended at public hospitals and 20 (14%) were attended at private centres, although 2 of the latter group had previously consulted their family doctor through the public healthcare system. Of the patients attended at public centres, 16 had occasionally consulted at private healthcare centres. Therefore, 25% of patients consulted through the private system at some point in the diagnostic pathway.
The mean diagnostic delay was 13.1 months (median, 11.7) after symptom onset, with riluzole treatment being introduced after a mean of 13.3 months (median, 12.2). The time taken to diagnose ALS can be divided into 3 diagnostic periods: the time between symptom onset and the first consultation; the time between the first consultation and first visit to a neurologist; and the time between first consultation with a neurologist and the diagnosis of ALS (Fig. 2).

Diagnostic pathway and delay: public vs private healthcare systems. Diagnostic pathways were similar under both healthcare systems, although patients attended at private centres consulted later and saw neurologists sooner due to the specific characteristics of that system. However, the total diagnostic delay was similar in both systems, as neurologists in the private system took longer to reach a diagnosis.
In our sample, the mean duration of the first phase was 3.6 months (median, 2.5), with the first physician (usually the family doctor) managing the case correctly (referral to a neurologist within one month) in 38% of cases.
The time between the initial medical consultation and the first visit to a neurologist was highly variable, with a mean duration of 4.48 months (median, 2.8). A neurologist was the first specialist consulted in 53% of cases. Despite this, 29% of patients were referred to other specialists after assessment by a neurologist; therefore, 65% of all patients were examined by a specialist other than a neurologist at some point prior to diagnosis, with traumatology (26%) and otorhinolaryngology (20%) being the most frequent referrals. As a result, 50% of patients underwent tests that do not inform the diagnosis of ALS (radiography, endoscopy, etc.), with 5% undergoing surgery related to the initial symptoms (normally for cervical or lumbar disc herniation).
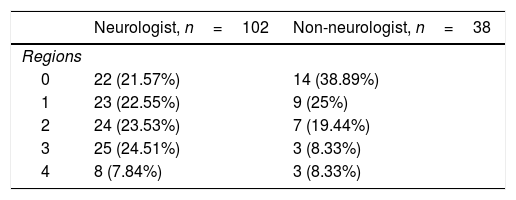
Neurologists diagnosed ALS after a mean time of 5.2 months and 3 consultations, with an initial misdiagnosis in 36% of cases. This was despite the fact that EP study findings were already available for 23% of patients at the time of consultation. Therefore, a large part of the diagnostic delay takes place after the first EP study (Table 1). In fact, half of the patients underwent more than one EP study before the diagnosis was established, as these studies were inconclusive in 34% of cases, most frequently (26%) because they did not meet Awaji criteria for involvement of any region. Furthermore, the first electromyography (EMG) only found a single region to be affected in 24% of patients. The first EP study was less informative (showing fewer regions affected according to the Awaji criteria) when it was not ordered by a neurologist (odds ratio, 0.7 [0.50-0.96]; P=.034; Table 2).
Number of regions affected (Awaji criteria), according to whether the electrophysiological study was ordered by a neurologist.
| Neurologist, n=102 | Non-neurologist, n=38 | |
|---|---|---|
| Regions | ||
| 0 | 22 (21.57%) | 14 (38.89%) |
| 1 | 23 (22.55%) | 9 (25%) |
| 2 | 24 (23.53%) | 7 (19.44%) |
| 3 | 25 (24.51%) | 3 (8.33%) |
| 4 | 8 (7.84%) | 3 (8.33%) |
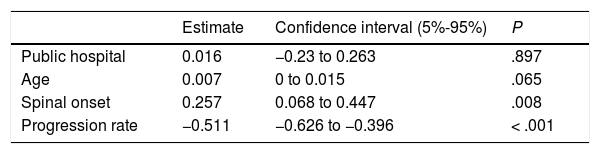
Most patients treated within the public healthcare system reported their initial symptoms to their family doctor, whereas those attended at private centres mainly consulted directly with specialists. Patients attended at private healthcare centres took slightly longer to seek medical care, perhaps due to slower symptom progression. Despite this, the more direct access to specialists meant that the mean time until consultation with a neurologist was one month shorter in this group. However, the diagnostic delay was similar in both systems after adjusting for age, form of onset, and progression rate (P=.897; Table 3). This may be due to the fact that, compared to the public system, patients assessed at private centres more frequently visited specialists other than neurologists or underwent a second EP study before diagnosis was established, resulting in longer times between the first EMG study and diagnosis (Table 1). Besides this, the diagnostic pathway and the impact in terms of unnecessary tests and non-indicated interventions were similar in both healthcare systems (Table 1 and Fig. 2).
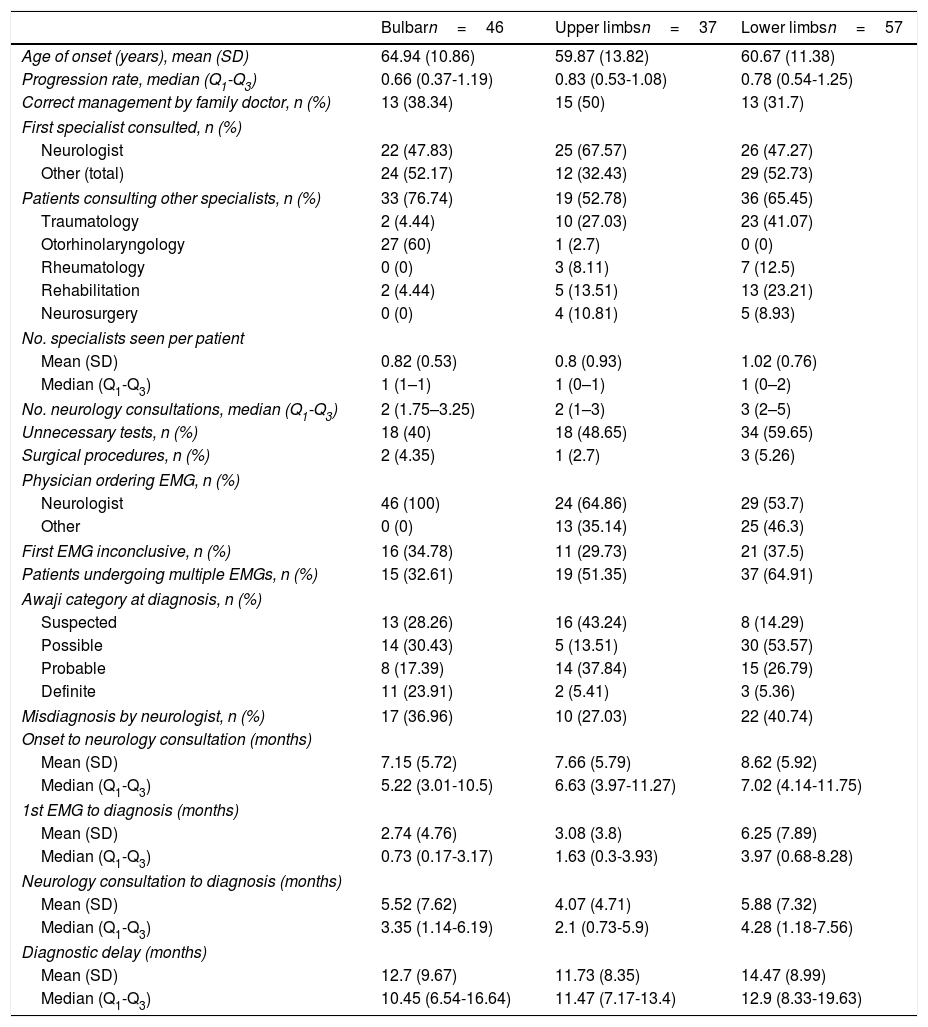
Diagnostic pathway according to region of onsetAccording to our analytical model (Table 3), diagnostic delay was greater in patients with spinal-onset ALS, independently of age, progression rate, or healthcare system. This difference may be attributed to the greater number of unnecessary tests performed in these patients (55% vs 40% for those with bulbar onset) and to the greater number of patients undergoing multiple EMG studies. Furthermore, the time between the first EMG study and diagnosis was longer in patients with spinal onset than among those with bulbar onset, despite the fact that similar percentages of patients with bulbar (30%) and with lower-limb onset (30%) showed no EMG alterations (Awaji criteria); this percentage was lower among patients with upper-limb onset (19%).
The longer delay in patients with spinal-onset ALS may be attributed to those with onset in the lower limbs. As shown in Table 4, these patients presented the poorest management by the family doctor and consequently saw neurologists later than patients in other groups. These patients were also the most difficult to diagnose, both for the neurologist and for other specialists, hence the higher numbers of unnecessary tests ordered and misdiagnoses. It is also common in these cases for the EMG not to be ordered by a neurologist; consequently, a second study is more frequently needed, resulting in longer delays and more consultations before ALS is diagnosed.
Diagnostic pathway according to region of onset.
| Bulbarn=46 | Upper limbsn=37 | Lower limbsn=57 | |
|---|---|---|---|
| Age of onset (years), mean (SD) | 64.94 (10.86) | 59.87 (13.82) | 60.67 (11.38) |
| Progression rate, median (Q1-Q3) | 0.66 (0.37-1.19) | 0.83 (0.53-1.08) | 0.78 (0.54-1.25) |
| Correct management by family doctor, n (%) | 13 (38.34) | 15 (50) | 13 (31.7) |
| First specialist consulted, n (%) | |||
| Neurologist | 22 (47.83) | 25 (67.57) | 26 (47.27) |
| Other (total) | 24 (52.17) | 12 (32.43) | 29 (52.73) |
| Patients consulting other specialists, n (%) | 33 (76.74) | 19 (52.78) | 36 (65.45) |
| Traumatology | 2 (4.44) | 10 (27.03) | 23 (41.07) |
| Otorhinolaryngology | 27 (60) | 1 (2.7) | 0 (0) |
| Rheumatology | 0 (0) | 3 (8.11) | 7 (12.5) |
| Rehabilitation | 2 (4.44) | 5 (13.51) | 13 (23.21) |
| Neurosurgery | 0 (0) | 4 (10.81) | 5 (8.93) |
| No. specialists seen per patient | |||
| Mean (SD) | 0.82 (0.53) | 0.8 (0.93) | 1.02 (0.76) |
| Median (Q1-Q3) | 1 (1–1) | 1 (0–1) | 1 (0–2) |
| No. neurology consultations, median (Q1-Q3) | 2 (1.75–3.25) | 2 (1–3) | 3 (2–5) |
| Unnecessary tests, n (%) | 18 (40) | 18 (48.65) | 34 (59.65) |
| Surgical procedures, n (%) | 2 (4.35) | 1 (2.7) | 3 (5.26) |
| Physician ordering EMG, n (%) | |||
| Neurologist | 46 (100) | 24 (64.86) | 29 (53.7) |
| Other | 0 (0) | 13 (35.14) | 25 (46.3) |
| First EMG inconclusive, n (%) | 16 (34.78) | 11 (29.73) | 21 (37.5) |
| Patients undergoing multiple EMGs, n (%) | 15 (32.61) | 19 (51.35) | 37 (64.91) |
| Awaji category at diagnosis, n (%) | |||
| Suspected | 13 (28.26) | 16 (43.24) | 8 (14.29) |
| Possible | 14 (30.43) | 5 (13.51) | 30 (53.57) |
| Probable | 8 (17.39) | 14 (37.84) | 15 (26.79) |
| Definite | 11 (23.91) | 2 (5.41) | 3 (5.36) |
| Misdiagnosis by neurologist, n (%) | 17 (36.96) | 10 (27.03) | 22 (40.74) |
| Onset to neurology consultation (months) | |||
| Mean (SD) | 7.15 (5.72) | 7.66 (5.79) | 8.62 (5.92) |
| Median (Q1-Q3) | 5.22 (3.01-10.5) | 6.63 (3.97-11.27) | 7.02 (4.14-11.75) |
| 1st EMG to diagnosis (months) | |||
| Mean (SD) | 2.74 (4.76) | 3.08 (3.8) | 6.25 (7.89) |
| Median (Q1-Q3) | 0.73 (0.17-3.17) | 1.63 (0.3-3.93) | 3.97 (0.68-8.28) |
| Neurology consultation to diagnosis (months) | |||
| Mean (SD) | 5.52 (7.62) | 4.07 (4.71) | 5.88 (7.32) |
| Median (Q1-Q3) | 3.35 (1.14-6.19) | 2.1 (0.73-5.9) | 4.28 (1.18-7.56) |
| Diagnostic delay (months) | |||
| Mean (SD) | 12.7 (9.67) | 11.73 (8.35) | 14.47 (8.99) |
| Median (Q1-Q3) | 10.45 (6.54-16.64) | 11.47 (7.17-13.4) | 12.9 (8.33-19.63) |
On the other hand, cases of upper-limb onset seem to be more easily diagnosed. This is demonstrated by the fact that these patients were most often managed correctly by family doctors, with less frequent referrals to specialists other than neurologists. Once they were seen by neurologists, they were also diagnosed sooner and showed a lower rate of misdiagnosis.
It is worth noting that the other specialties consulted varied according to the region of onset; patients with bulbar onset were more frequently referred to otorhinolaryngologists, and those with spinal onset more frequently visited traumatologists.
DiscussionAlmost 20 years after the publication of the revised El Escorial criteria, early diagnosis of ALS remains an outstanding problem, as shown by numerous studies conducted in different countries. This study shows that our setting is no exception to this rule.
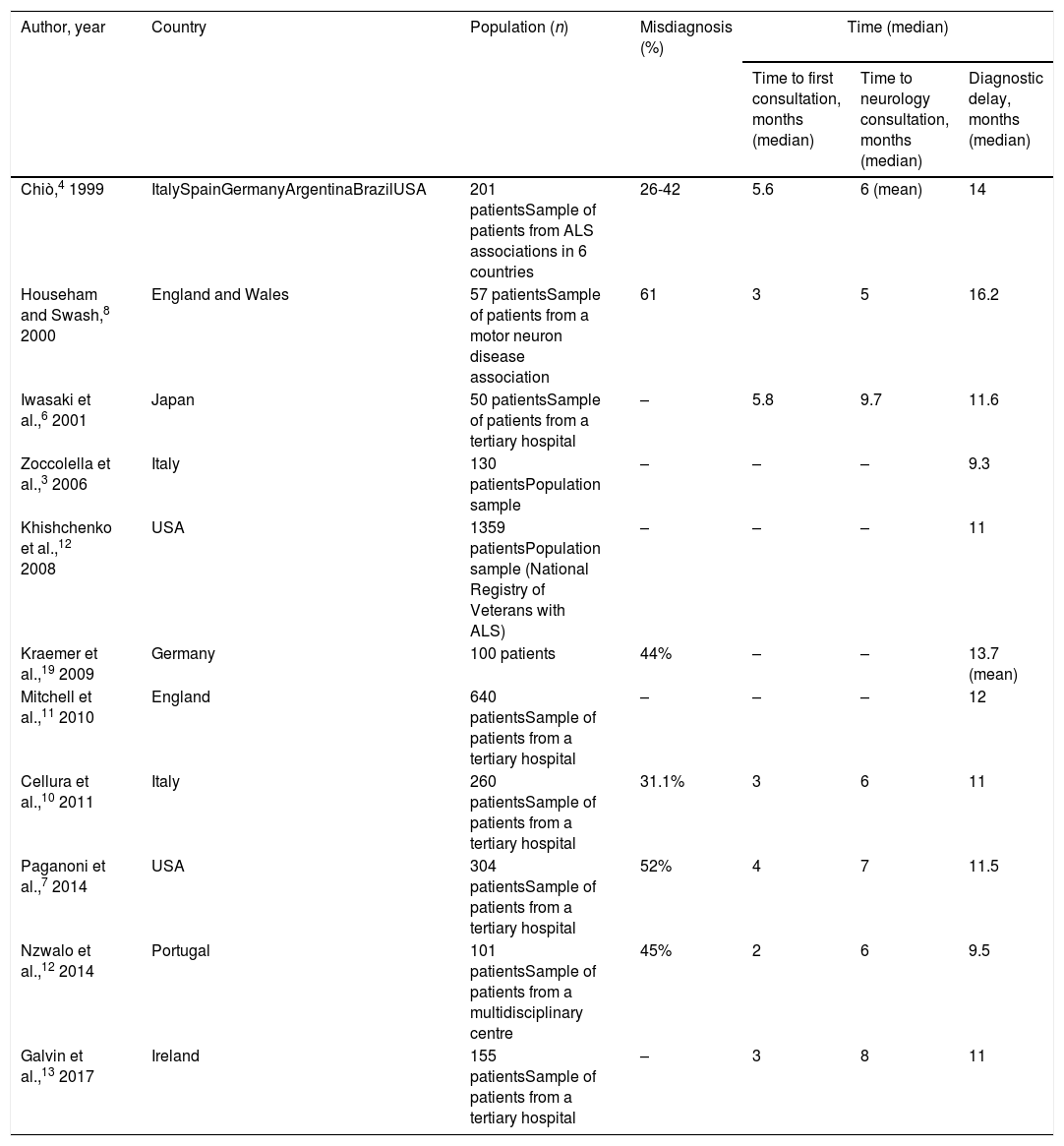
The appearance of symptoms of ALS represents the beginning of a long journey for patients and their families, with diagnosis taking approximately 12 months according to our results and to previous studies (Table 5). This pre-diagnostic period typically comprises several stages of roughly equal duration; therefore, diagnostic delay cannot be attributed to shortcomings in any single stage, but rather is probably explained by multiple factors in each stage. These are briefly addressed below.
Diagnostic delay for amyotrophic lateral sclerosis according to previous studies.
| Author, year | Country | Population (n) | Misdiagnosis (%) | Time (median) | ||
|---|---|---|---|---|---|---|
| Time to first consultation, months (median) | Time to neurology consultation, months (median) | Diagnostic delay, months (median) | ||||
| Chiò,4 1999 | ItalySpainGermanyArgentinaBrazilUSA | 201 patientsSample of patients from ALS associations in 6 countries | 26-42 | 5.6 | 6 (mean) | 14 |
| Househam and Swash,8 2000 | England and Wales | 57 patientsSample of patients from a motor neuron disease association | 61 | 3 | 5 | 16.2 |
| Iwasaki et al.,6 2001 | Japan | 50 patientsSample of patients from a tertiary hospital | – | 5.8 | 9.7 | 11.6 |
| Zoccolella et al.,3 2006 | Italy | 130 patientsPopulation sample | – | – | – | 9.3 |
| Khishchenko et al.,12 2008 | USA | 1359 patientsPopulation sample (National Registry of Veterans with ALS) | – | – | – | 11 |
| Kraemer et al.,19 2009 | Germany | 100 patients | 44% | – | – | 13.7 (mean) |
| Mitchell et al.,11 2010 | England | 640 patientsSample of patients from a tertiary hospital | – | – | – | 12 |
| Cellura et al.,10 2011 | Italy | 260 patientsSample of patients from a tertiary hospital | 31.1% | 3 | 6 | 11 |
| Paganoni et al.,7 2014 | USA | 304 patientsSample of patients from a tertiary hospital | 52% | 4 | 7 | 11.5 |
| Nzwalo et al.,12 2014 | Portugal | 101 patientsSample of patients from a multidisciplinary centre | 45% | 2 | 6 | 9.5 |
| Galvin et al.,13 2017 | Ireland | 155 patientsSample of patients from a tertiary hospital | – | 3 | 8 | 11 |
Patients took approximately 3 months to seek medical attention after symptoms presented; previous studies report similar findings (Table 5). However, a significant percentage of patients (14% in our sample) consulted within 15 days of onset. While we may attribute this to patients’ failure to accurately recall the exact date of symptom onset, in at least some cases we were able to verify acute onset of symptoms in the form of pain or following surgery, which appears to have acted as a trigger factor. Previous studies report that up to one-third of patients with ALS present pain before or concurrently with symptom onset18; this finding is noteworthy as, according to our results, these appear to be the patients who consult early. However, this form of onset is not necessarily associated with reduced diagnostic delay, probably because pain is considered an atypical symptom of ALS and is suggestive of other diagnoses. Regarding the role of surgery as a trigger factor, one study found that 3.5% of patients with ALS had undergone procedures in the 3 months prior to symptom onset, with an association between the location of the procedure and the region of symptom onset; disease progression also appeared to accelerate after surgery.19 These findings support the hypothesis that surgery may play a pathogenic role.
First consultation to neurology consultationAll studies report the duration of this period at approximately 4 months (Table 5). Rather than waiting lists, this delay seems to be explained mainly by incorrect working diagnoses established by the family doctor. Neurological disease was suspected early in only 38% of patients, and approximately half of patients were referred to other specialists (mainly otorhinolaryngologists for cases of bulbar onset and traumatologists for spinal onset) before seeing a neurologist. However, these results are better than those reported in another European study.13
Neurology consultation to diagnosisThis period may be the most variable between patients (mean, 5.2 months; median, 2.6). In any case, we consider this delay to be excessive, particularly given the period of almost 8 months between symptom onset and first consultation with a neurologist. As in other studies (Table 5), the diagnosis established in the initial neurology consultation was incorrect in over one-third of cases; many patients were also referred to other specialists, increasing diagnostic delay. The delay cannot be attributed to failure to promptly perform the EP study, as the study was performed early in this period. In fact, most patients underwent multiple studies before a diagnosis could be reached.
Factors modifying diagnostic delayAs is the case with diagnostic delay and the different stages of the diagnostic process, misdiagnosis is constantly reported in various studies, independently of cultural context and healthcare system characteristics (Table 5). In fact, our results show no difference in diagnostic delay between patients assessed in public and in private healthcare centres, despite differences between the systems, with more direct access to specialists (without mediation by the family doctor) and shorter waits for complementary tests in the private system. Patients assessed under the private system generally saw neurologists and underwent EMG testing sooner. However, this did not translate to a reduction in diagnostic delay, as the time from the first neurology consultation and EMG study to diagnosis was longer in these patients. This suggests, firstly, that diagnostic delay cannot be attributed to weaknesses specific to the healthcare system, such as waiting lists, and secondly, that early consultation with a neurologist does not guarantee early diagnosis, as other authors have noted.8,10,13
Bulbar onset and rapid progression are clearly associated with shorter diagnostic delay3,6,12,20,21; this finding was replicated in our study. Rapid progression not only leads to earlier consultation, but also facilitates diagnosis of ALS, as continuous, observable progression is a requirement for diagnosis. Bulbar-onset ALS merits specific discussion. While some authors attribute the reduced diagnostic delay in these patients to faster progression,12 our findings suggest that this effect is independent of progression rate and can largely be explained by the greater delays associated with lower-limb onset. These patients are more likely to be misdiagnosed and to undergo unnecessary tests as, to the untrained eye, ALS mimics a broad range of highly prevalent diseases, and especially traumatological conditions (disc herniation, degenerative bone disease, etc.); this phenomenon is much more prevalent in cases of bulbar or upper-limb onset.
Impact of diagnostic delay on patients and the healthcare systemOur results are consistent with those of previous studies, and show that misdiagnosis has a considerable negative impact not only on patients (and their families),7,13 but also on the healthcare system, with an estimated cost of €3500 per patient.13 Of this amount, €2000 could be saved if the diagnostic pathway followed the recommendations of the current guidelines.2
How to shorten diagnostic delaysAs misdiagnosis is often explained in large measure by the disease's insidious onset and considerable clinical heterogeneity, one possible course of action could be to provide special training to the specialists (family doctors, otorhinolaryngologists, traumatologists) involved in the diagnostic pathway of patients with ALS, placing particular emphasis on the so-called red flags. However, as a family doctor will probably see only a handful of cases of ALS over the course of his/her career, it is unlikely that this measure would have a significant effect on diagnostic delay (as has been shown in previous studies),13 particularly given the fact that earlier consultation with a neurologist does not guarantee faster diagnosis.8,10,13
Neither does diagnostic delay appear to be affected by long waiting lists, as tests are frequently expedited when ALS is suspected, with some centres even scheduling hospital admissions. Furthermore, previous attempts to implement fast referral systems have not achieved significant reductions in diagnostic delay.11
Therefore, action to reduce diagnostic delay seems most promising in the period after the first neurology consultation.
The El Escorial criteria and their subsequent revisions have clearly failed in the aim of facilitating earlier diagnosis. Therefore, and given the absence of specific markers, diagnosis of ALS continues to be clinical, and depends fundamentally on the experience of the neurologist. EP studies constitute an invaluable tool, provided that the study is targeted in line with clinical suspicion and is correctly interpreted. If this is not the case, the study may not be optimal and results may be inconclusive, leading to confusion, misdiagnosis, and repeated testing. It should be noted that in over one-third of cases, the first EP study was not compatible with the diagnosis of motor neuron disease (Awaji criteria15), mainly because the results did not fulfil criteria for involvement of any specific region. This is consistent with the results of previous studies,22 and demonstrates how the El Escorial electrophysiological criteria and the subsequent revised (Awaji) criteria,15,23 designed for research use, should not be used in clinical practice, especially in the pursuit of early diagnosis. We should be mindful that certain phenotypes, such as progressive bulbar and pseudobulbar palsy, typically show normal EMG results in the early months or years of the disease, and that up to 20% of patients with ALS may present decreased sensory action potential amplitude in the EP study.24 As a result, an EMG study returning normal or atypical findings is not incompatible with the diagnosis of motor neuron disease if clinical suspicion is well founded. Therefore, important measures in reducing diagnostic delay may include specific training for general neurologists to diagnose motor neuron conditions, or referral to reference centres for patients in whom diagnostic suspicion cannot be confirmed.
ConclusionsOur findings show a similar diagnostic pathway and delay to those reported in other European countries; rather than being affected by the peculiarities of each healthcare system, they appear to be inherent to the disease, and have a negative impact both for patients and for healthcare systems. Lower-limb onset represents the greatest diagnostic challenge due to the broad range of differential diagnoses. While there is room for improvement at every stage of the diagnostic pathway, misdiagnosis by the neurologist is frequent, and may partially be attributed to an incorrect working diagnosis or interpretation of the EP study. Therefore, 2 measures that may assist in reducing diagnostic delays are specific training for general neurophysiologists and neurologists, and referral to reference centres. Future studies should analyse whether early referral to reference centres reduces diagnostic delay.
FundingJFVC is funded by a research contract with the La Fe Healthcare Research Institute (2016/0490).
Conflicts of interestNone declared.
The following are the supplementary data to this article:
Please cite this article as: Vázquez-Costa JF, Martínez-Molina M, Fernández-Polo M, Fornés-Ferrer V, Frasquet-Carrera M, Sevilla-Mantecón T. Análisis del trayecto y retraso diagnóstico de los pacientes con esclerosis lateral amiotrófica en la Comunidad Valenciana. Neurología. 2021;36:504–513.
