



La angiopatía amiloide cerebral, esto es, el depósito de β-amiloide, fundamentalmente Aβ40, en la pared de las pequeñas arterias cerebrales y leptomeníngeas, constituye un hallazgo muy frecuente en la enfermedad de Alzheimer y en sujetos cognitivamente sanos de edad avanzada1–3. El diagnóstico definitivo es anatomopatológico, aunque la presencia de cambios típicos en la RM cerebral, junto con la demostración de patología amiloide cerebral mediante PET-PIB o midiendo el descenso de Aβ-40 y Aβ-42 en líquido cefalorraquídeo (LCR), permite obviar la histopatología3. Una forma infrecuente de presentación es la denominada angiopatía amiloide inflamatoria (AAI), que se caracteriza por la aparición de un infiltrado inflamatorio perivascular asociado a la angiopatía amiloide y que suele cursar con un cuadro clínico que combina deterioro cognitivo rápidamente progresivo, crisis epilépticas y cefalea. Esta entidad se describió en los años ochenta como un tipo de vasculitis aislada del sistema nervioso central (SNC), si bien con los primeros casos de meningoencefalitis inducida por la vacuna Aβ pasó a considerarse una entidad diferente en la que el β-amiloide depositado en la pared de los vasos actúa como antígeno4–6. Esta hipótesis se ha visto reforzada con la descripción de anticuerpos frente Aβ-42 en el LCR y el parénquima cerebral, y por los nuevos estudios histopatológicos7–9.
Presentamos el caso de un paciente con AAI y enfermedad de Alzheimer en el que consideramos que fue prescindible la biopsia para llegar al diagnóstico.
Paciente varón, de 82 años, que acude a urgencias tras presentar el día del ingreso un traumatismo craneoencefálico por una caída. Como antecedente de interés el paciente había presentado múltiples epiteliomas basocelulares faciales extirpados quirúrgicamente e irradiados en el 2003 por recidiva local. En urgencias se encuentra al paciente desorientado y sin focalidad neurológica. La TC mostraba focos hemorrágicos y amplias áreas hipodensas en la sustancia blanca bihemisférica, que sugerían una neoplasia subyacente, ingresando en neurocirugía con tratamiento con dexametasona 4mg/8h.
Unos 6 meses antes del ingreso, el paciente había comenzado a tener despistes; 3 meses después había desarrollado apatía, temblor en las manos y desorientación. La semana previa al ingreso había empeorado notablemente y necesitaba supervisión para ducharse.
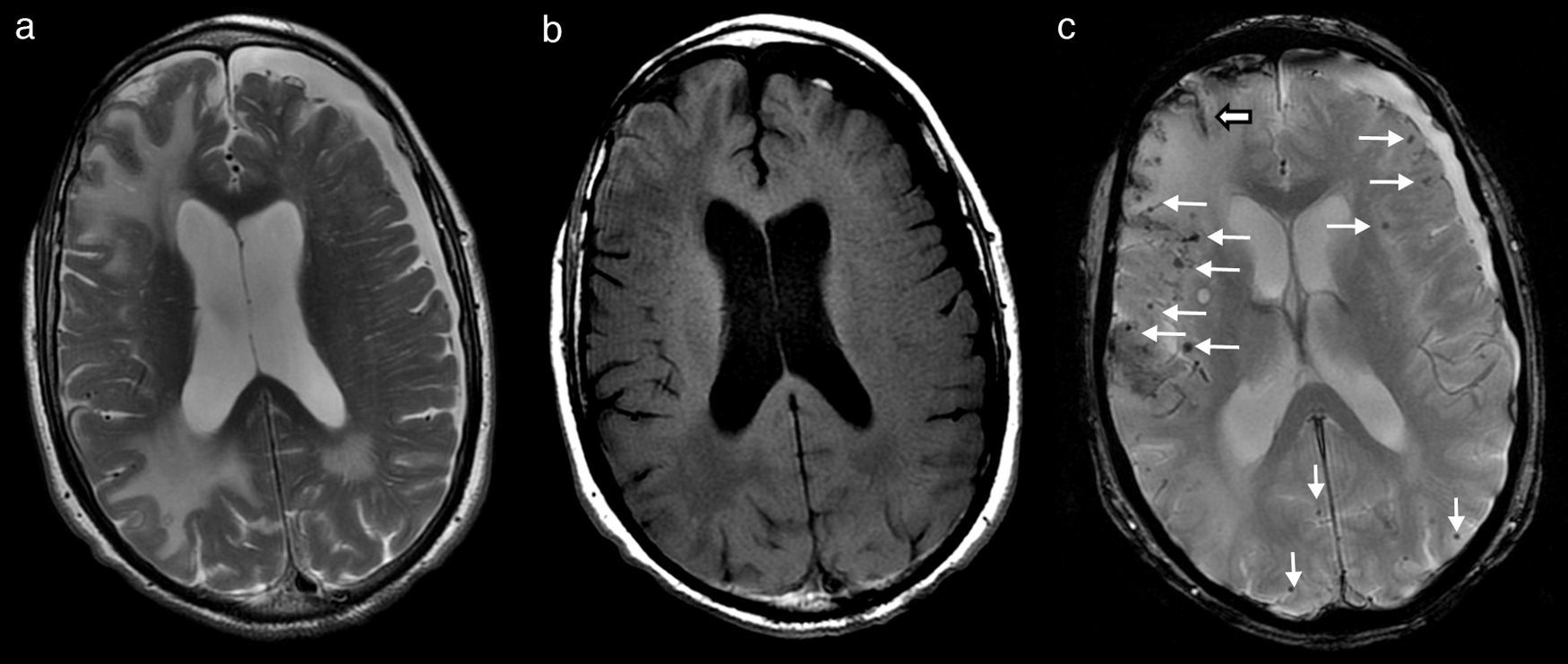
Durante los primeros días del ingreso, empeoró su función cognitiva, con episodios de agitación y alucinaciones nocturnas. No obstante, a la semana del ingreso, en tratamiento con dexametasona, comienzan a mejorar de forma significativa tanto el temblor como su función cognitiva, hasta el punto de que la familia lo encontraba mejor que antes del ingreso. En ese momento se realiza una RM cerebral que muestra amplias áreas de edema vasogénico occipitoparietal y frontal derecho, y de forma más limitada izquierdo (fig. 1a y b), así como múltiples focos de depósito siderótico milimétricos, de predominio cortical difusos (fig. 1c). Ante los hallazgos, se consulta con neurología.
 e hipointensas en T1 (b), correspondientes a amplias áreas de edema vasogénico subcortical occipitoparietal y frontal derecho, y de forma más limitada parietooccipital izquierdo. En secuencia eco de gradiente potenciada en T2* (c) se observan múltiples focos de depósito siderótico milimétricos, de predominio cortical difusos (flechas finas) y restos de siderosis leptomeníngea subaracnoidea (flecha gruesa). Existe una atrofia corticosubcortical con marcada dilatación involutiva de espacios perivasculares de Virchow-Robin y un higroma subdural frontoparietal izquierdo.")
RM cerebral que muestra lesiones hiperintensas en T2 (a) e hipointensas en T1 (b), correspondientes a amplias áreas de edema vasogénico subcortical occipitoparietal y frontal derecho, y de forma más limitada parietooccipital izquierdo. En secuencia eco de gradiente potenciada en T2* (c) se observan múltiples focos de depósito siderótico milimétricos, de predominio cortical difusos (flechas finas) y restos de siderosis leptomeníngea subaracnoidea (flecha gruesa). Existe una atrofia corticosubcortical con marcada dilatación involutiva de espacios perivasculares de Virchow-Robin y un higroma subdural frontoparietal izquierdo.
En ese momento, el examen neurológico muestra un marcado parkinsonismo bilateral y simétrico, una desorientación parcial en las 3 esferas, un importante déficit de atención y una moderada afectación de todas las funciones cognitivas. En el Minimental puntúa 13/30.
Establecido el diagnóstico sindrómico de demencia subaguda, ante los hallazgos de la exploración y de la RM cerebral, se plantea el diagnóstico diferencial de la leucoencefalopatía, fundamentalmente entre una AAI, una vasculitis aislada del SNC, una por radionecrosis, un linfoma primario del SNC y una leucoencefalopatía multifocal progresiva. En consecuencia, se realizan bioquímica, hemograma, vitamina B12, hormonas tiroideas, estudio de autoinmunidad y serologías del virus de la inmunodeficiencia humana y la sífilis, resultando todas ellas normales. Se realiza una punción lumbar que muestra una discreta proteinorraquia (0 células, glucosa: 61mg/dl, proteínas 56mg/dl), con un índice de IgG normal y ausencia de bandas oligoclonales. Se determinó la concentración en el LCR de Aβ-42 (251 pg/ml [> 500pg/ml]), de fosfo-Tau (53pg/ml [< 85pg/ml]) y de h-Tau (1.520 pg/mL [< 350pg/ml]).
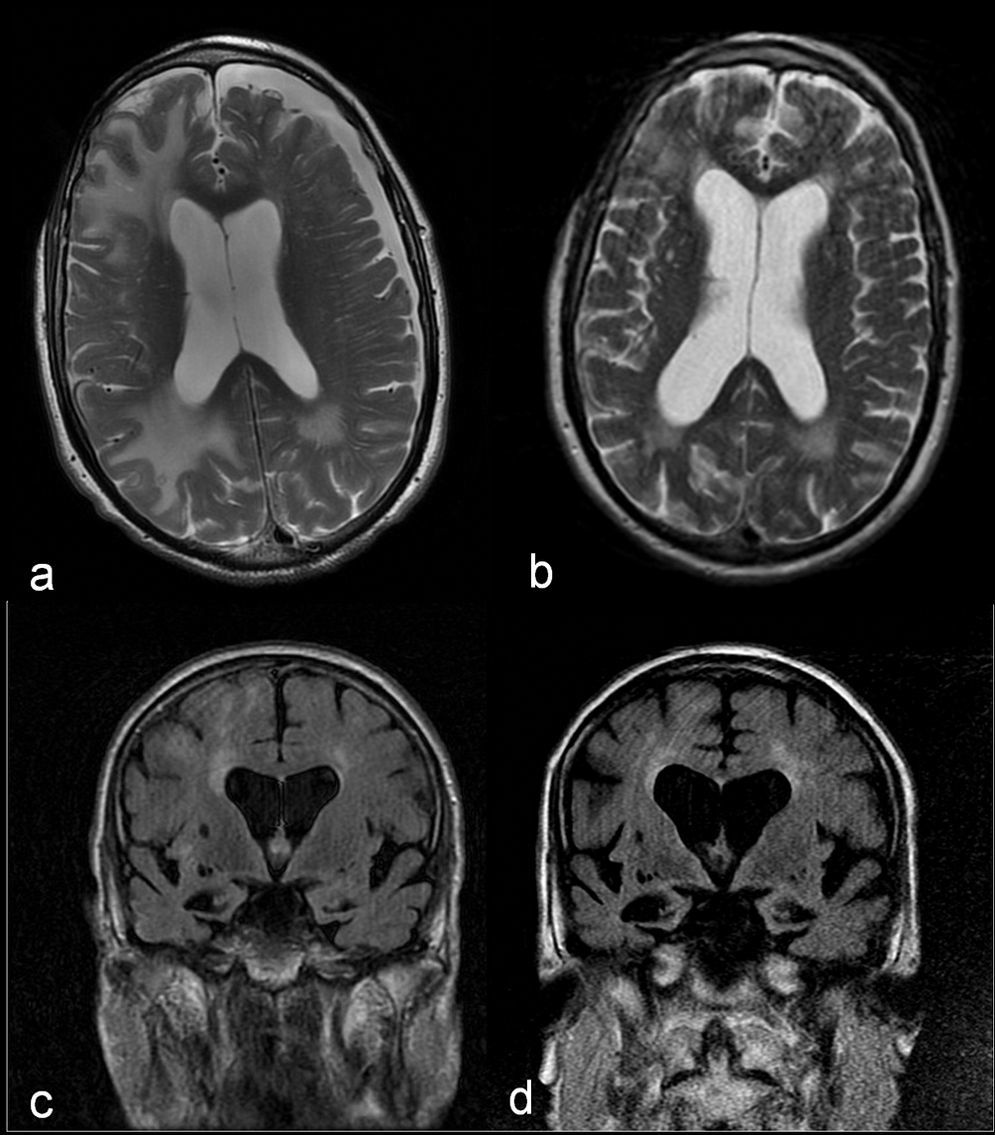
Estos resultados, unidos a la buena respuesta inicial con dexametasona, plantean como primera opción diagnóstica una AAI, por lo que se desestima practicar una biopsia y se opta por administrar un bolo de 1g de 6-metilprednisolona por vía intravenosa durante 5 días, con posterior pauta descendente de prednisona, evolucionando a una mejoría progresiva en su domicilio y recuperación de actividades previas. Un mes y medio después del inicio del cuadro se realiza una nueva RM cerebral, que muestra una notable disminución del tamaño de las lesiones en la sustancia blanca (fig. 2a y b). No obstante, cada intento de reducir la dosis de prednisona por debajo de 10mg/día empeoraba la función cognitiva, con recuperaciones parciales al aumentar la dosis. A los 7 meses del alta, la RM cerebral muestra estabilidad de las lesiones y progresión de la atrofia de hipocampos (fig. 2c y d). En el plano cognitivo ha presentado un lento deterioro (Minimental 11/30). Se establece el diagnóstico de probable AAI y probable enfermedad de Alzheimer.
 y un mes y medio después (b). Imágenes comparativas en FLAIR coronal que muestran la progresión de la atrofia cerebral y de hipocampos, y la dilatación ventricular secundaria entre la RM cerebral realizada en el momento agudo (c) y 6 meses después (d).")
Imágenes comparativas en secuencia T2 axial donde se observa una notable disminución en el tamaño de las lesiones de la sustancia blanca y resolución del higroma subdural entre la RM cerebral realizada en el momento agudo (a) y un mes y medio después (b). Imágenes comparativas en FLAIR coronal que muestran la progresión de la atrofia cerebral y de hipocampos, y la dilatación ventricular secundaria entre la RM cerebral realizada en el momento agudo (c) y 6 meses después (d).
El diagnóstico de la AAI resulta inequívoco si existe confirmación histológica, si bien cada vez son más los autores que propugnan un diagnóstico basado en un cuadro clínico-radiológico compatible (lesiones parcialmente reversibles hiperintensas en T2/FLAIR y mapa ADC e hipointensas en T1, con ocasional captación de contraste y microhemorragias visibles en eco de gradiente), con exclusión de otras leucoencefalopatías y apoyado por algún marcador de depósito amiloide (p. ej., descenso de Aβ-42 en el LCR)1,7,8,10–13. El tratamiento en la fase aguda consiste en megadosis de corticoides, con posterior pauta descendente. Al menos el 25% no responde a corticoides o presenta un curso recidivante, pudiendo responder a inmunosupresores10,14.
En nuestro caso, una cierta demora en el tratamiento desde el inicio de la clínica pudo haber propiciado que la recuperación no fuera completa tras el tratamiento. Por otro lado, el curso lentamente progresivo, pese al tratamiento con corticoides, unido a la presencia de una significativa y progresiva atrofia cerebral y de hipocampos hace suponer la presencia de enfermedad de Alzheimer asociada a la AAI15.
Este caso fue presentado en el concurso de casos clínicos de la Sociedad Valenciana de Neurología.

