



Cerebral folate deficiency (CFD) is a neurological syndrome associated with low concentrations of 5-methyltetrahydrofolate in the cerebrospinal fluid (CSF) despite normal serum folate levels.1 It may present at any age, from the prenatal period to adulthood, and is associated with a wide range of phenotypes.2 The generation of autoantibodies against folate receptors,3 pathogenic variants of the folate receptor alpha (FOLR1) gene, and mitochondrial dysfunction have been reported as causes of CFD.2,4 However, other mechanisms may also be involved, since the condition has been reported in patients with a wide range of neurological and psychiatric disorders.5,6
We present the case of a patient with CFD who was subsequently diagnosed with hereditary spastic paraplegia.
Our patient was a 26-year-old man who, at the age of 10 years, presented progressive cognitive and behavioural alterations, difficulty walking, and generalised tremor starting in the left arm.
When he was first examined, at 16 years of age, he presented multiple neurological symptoms, including cognitive impairment, intrusive saccades, movement disorder (multifocal dystonia, generalised tremor, and bradykinesia), pyramidal syndrome, and gait ataxia with pes cavus.
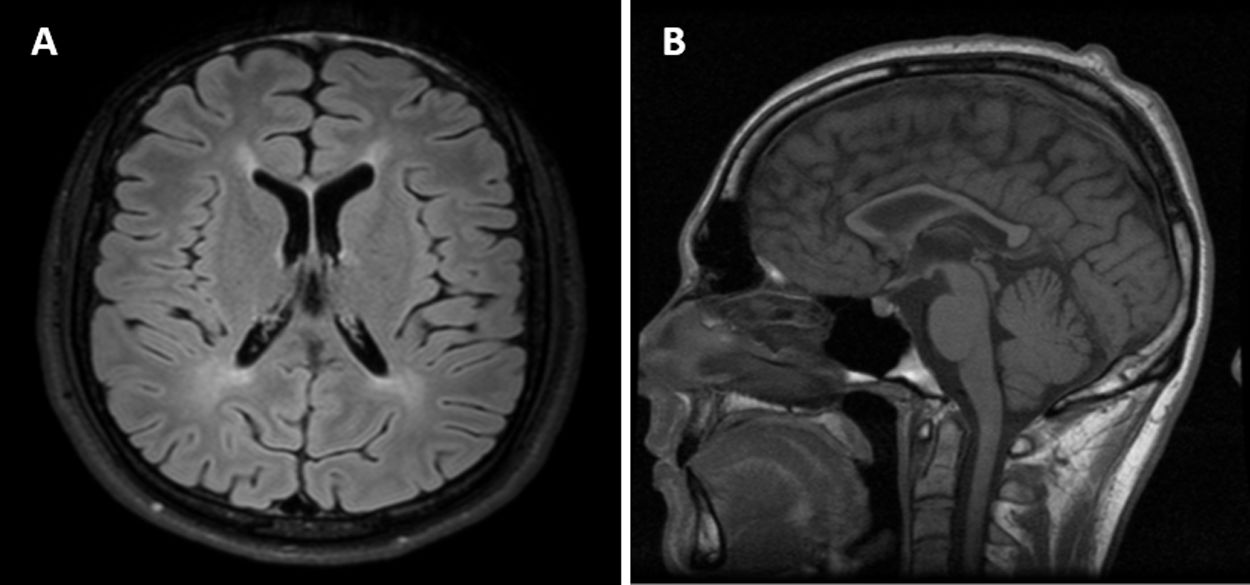
The neuropsychological examination revealed frontostriatal dysfunction, and electroencephalography (EEG) detected slow background activity (6 Hz). Brain MRI revealed a hyperintensity in the periventricular white matter and global atrophy, with marked atrophy of the corpus callosum (Fig. 1).
 Axial T2-FLAIR sequence showing periventricular white matter hyperintensity. B) T1-weighted sequence showing global atrophy, with marked atrophy of the corpus callosum.")
Extrapyramidal symptoms improved considerably with levodopa.
We subsequently identified CFD (CSF folate level < 20 nmol/L; normal range, 50-78), with normal levels of neurotransmitters, pterins, and amino acids. Serum folate levels were within the normal range. We ruled out the presence of variants in genes FOLR1 and POLG, and in the mitochondrial DNA. Analysis of autoantibodies against folate receptors in the serum revealed elevated titres of blocking antibodies (4.77 pmol of folate receptor blocked per mL serum).
Folinic acid supplementation (30 mg/day) normalised CSF folate levels and significantly improved cognitive function, extrapyramidal symptoms, and oculomotor and EEG alterations, but had no effect on pyramidal signs.
At age 18, spastic paraparesis was evident. The patient is now unable to stand or walk unassisted. Other neurological signs, such as parkinsonism and cognitive dysfunction, resolved nearly completely despite progressive decreases in the dosage of levodopa and anticholinergic medications. A brain and spinal cord MRI study ruled out new structural lesions, and CSF 5-methyltetrahydrofolate levels remained normal. The patient underwent 3 electrophysiological studies, at ages 19 and 24, which ruled out polyneuropathy and myopathy.
More recently, clinical exome sequencing identified 2 variants in the ZFYVE26 gene, c.1675 T > C (p.[Cys559Arg]) and c.3394C > T (p.[Gln1132*]), and BSCL2 gene variant c.1220C > T (p.[Pro407Leu]). ZFYVE26 gene variant c.3394C > T (p.[Gln1132*]), not previously reported, results in a truncated protein and has therefore been classified as pathogenic. The other ZFYVE26 variant is also a novel mutation, although its clinical significance is unknown; the same is true of the variant identified in BSCL2. Our patient’s parents, who were asymptomatic, also underwent genetic studies: the 2 ZFYVE26 variants were found in different alleles, and the BSCL2 gene variant was also detected in the father. The patient continued to receive folinic acid supplementation.
It has been hypothesised that the different phenotypes of CFD are determined by the age at which folate transport to the CNS is impaired.2 Initially localised and subsequently generalised dystonia, bradykinesia, and pyramidal syndrome have been described in patients with CFD presenting from adolescence to adulthood.2 Other characteristics include cognitive impairment and gait ataxia.2 This seems to be consistent with our patient’s symptoms.
Presence of elevated folate receptor autoantibody titres supports the association between CFD and our patient’s symptoms. Blocking antibodies have been detected in several conditions, including autism, schizophrenia, Rett syndrome, and Alpers disease.6,7
Progression of pyramidal syndrome justified clinical exome sequencing, identifying variants in genes ZFYVE26 and BSCL2, which are associated with autosomal recessive spastic paraplegia type 15 (SPG15) and autosomal dominant spastic paraplegia type 17 (SPG17), respectively. Our patient’s clinical phenotype resembles that of SPG15: cognitive impairment, behavioural alterations, pes cavus, ataxia, cerebral white matter hyperintensities on MRI, corpus callosum atrophy, and age of onset between 5 and 19 years.8 Although the BSCL2 variant was found in the father, who was asymptomatic, and this gene is associated with phenotypic variability and incomplete penetrance,9 our patient’s symptoms were not compatible with SPG17, and the variant is of uncertain significance.
As with other disorders, CFD may be associated with SPG15 and is probably responsible for some of our patient’s symptoms, considering the positive response to folinic acid supplementation, particularly in extrapyramidal symptoms. To our knowledge, this association has not previously been described, either at the clinical or the molecular level. We wonder whether some traits linked to SPG15 in previous reports (eg, parkinsonism10) may be associated with CFD. Our case suggests a possible association between these 2 entities, and underscores the need to consider CFD in patients with these disorders.
FundingThe authors have received no funding for this study.
We thank our patient and his family.
We also wish to thank Laura Vilarinho and Vasco Sá Pinto for their assistance in patient examination.
Please cite this article as: Duarte S, Cruz Martins R, Rodrigues M, Lourenço E, Moreira I, Alonso I, et al. Asociación de deficiencia cerebral de folato y paraplejía espástica hereditaria. Neurología. 2021;36:550–552.
