



Prionopathy is the cause of 62% of the rapidly progressive dementias (RPD) in which a definitive diagnosis is reached. The variability of symptoms and signs exhibited by the patients, as well as its different presentation, sometimes makes an early diagnosis difficult.
MethodsPatients with diagnosis of definite or probable prionopathy during the period 1999–2012 at our hospital were retrospectively reviewed. The clinical features and the results of the complementary tests (14-3-3 protein, EEG, MRI, FDG-PET, and genetic analysis) were evaluated in order to identify some factors that may enable an earlier diagnosis to be made.
ResultsA total of 14 patients are described: 6 with definite sporadic Creutzfeldt-Jakob (sCJD) disease, 3 with probable sCJD, 4 with fatal familial insomnia, and 1 with the new variant. The median age at diagnosis was 54 years old. The mean survival was 9.5 months. Mood disorder was the most common feature, followed by instability and cognitive impairment. 14-3-3 protein content in the cerebrospinal fluid was positive in 7 of 11 patients, and the EEG showed typical signs in 2 of 12 patients. Neuroimaging (FDG-PET, MRI) studies suggested the diagnosis in 13 of the 14 patients included.
ConclusionsMost patients presenting with RPD suffer from a prion disease. In our series the most useful complementary tests were MRI and FDG-PET, being positive in 13 of the 14 patients studied.
Las prionopatías representan hasta el 62% de los casos de demencia rápidamente progresiva (DRP) en los que se alcanza un diagnóstico definitivo. La variabilidad de los síntomas y signos iniciales y las diferencias en su evolución dificultan el diagnóstico precoz.
MétodosEstudio retrospectivo en el que se incluye a pacientes con prionopatía probable o definitiva, que acudieron a la consulta de Neurología de nuestro centro durante el periodo 1999–2012. Se describen las características clínicas y los resultados de las exploraciones complementarias (proteína 14-3-3, EEG, RM, PET-FDG y análisis genético), con la finalidad de identificar qué marcadores permiten un diagnóstico precoz.
ResultadosSe describe a 14 pacientes: 6 con enfermedad de Creutzfeldt-Jakob esporádica (ECJe) definitiva, 3 con ECJe probable, 4 con insomnio familiar fatal y uno con la nueva variante de la enfermedad de Creutzfeldt-Jakob. La mediana de edad al diagnóstico fue de 54 años y la mediana de supervivencia de 9,5 meses. El trastorno del ánimo fue el síntoma inicial más frecuente, seguido de inestabilidad de la marcha y deterioro cognitivo. La proteína 14-3-3 fue positiva en el líquido cefalorraquídeo en 7 de 11 pacientes y el EEG mostró signos típicos en 2 de 12 pacientes explorados. El estudio de neuroimagen mostró alteraciones en 13 de los 14 pacientes.
ConclusionesAdemás de la DRP, el trastorno conductual y de la marcha son síntomas iniciales frecuentes en las prionopatías. En nuestra serie, las pruebas complementarias más útiles para apoyar el diagnóstico fueron la RM y la PET-FDG.
Prion diseases constitute a group of neurodegenerative disorders caused by accumulation of PrPSc, an abnormal isoform of the cellular prion protein (PrPC).1 This cell-surface glycoprotein is made up of 209 amino acids and a disulphide bond.1 The abnormal isoform is infectious in the absence of nucleic acids.2 It is encoded by PRNP on chromosome 20, which presents a methionine (M)–valine (V) polymorphism at codon 129. Methionine homozygosity is a risk factor for developing prion diseases. They can be classified into acquired, hereditary, or sporadic forms.1 The annual incidence rate of prion diseases is approximately 1 case per million people.3 One of the most common clinical manifestations of these diseases is rapidly progressive dementia (RPD). In fact, up to 62% of patients with a form of RPD that can be conclusively diagnosed4 have some type of prion disease. However, since the first signs and symptoms vary greatly,5 only 18% of cases are diagnosed in the first assessment,6 and the correct diagnosis is usually assigned an average of 8 months after symptom onset.
The purpose of this study is to describe the clinical features of prion diseases and how certain supplementary tests can contribute to the diagnostic process. To this end, we have retrospectively studied the cases of prion disease registered at Clínica Universidad de Navarra (Navarre, Spain).
Patients and methodsWe conducted a retrospective study including all patients with either a probable or a definite diagnosis of prion disease, according to validated diagnostic criteria,7,8 who had been examined in the neurology department at Clínica Universidad de Navarra between 1999 and 2012. Clinical signs and symptoms and results from additional tests were collected from medical histories. As supplementary diagnostic tests, we used 14-3-3 protein in CSF, analysis of PRNP (study of known mutations and assessment of polymorphism at codon 129), and findings from electroencephalography and structural and functional neuroimaging studies. We visually analysed the presence or absence and the location of hyperintense regions in both diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences on MRI. We visually determined the presence or absence and the location of hypometabolic and/or hypoperfused areas in cerebral metabolism studies using PET with F-18 fluorodeoxyglucose (FDG-PET) in 13 patients, and using SPECT with Tc99m-hexamethylpropyleneamine oxime (99mTc-HMPAO SPECT) in another patient. For each additional test, each patient was assigned a pattern of impairment according to the presence and location of hyperintense, hypometabolic, and/or hypoperfused areas.7–9 We therefore established 3 different patterns: a cortical pattern showing hyperintensity, hypometabolism, and/or hypoperfusion in the cortex; a subcortical pattern characterised by hyperintensity, hypometabolism, and/or hypoperfusion in the basal ganglia and/or thalamus; and a cortical-subcortical pattern showing hyperintensity, hypometabolism, and/or hypoperfusion in the cortical and subcortical regions. MRI and FDG-PET findings were compared in order to assess the sensitivity of these 2 techniques. For all cases, we investigated whether a neuropathology study had been performed and checked those results.
ResultsClinical and epidemiological characteristicsWe detected some form of prion disease in 14 patients between 1999 and 2012. One patient had been diagnosed with variant Creutzfeldt-Jakob disease (vCJD); the diagnosis was later confirmed by an anatomical pathology study.7,10 Four patients were shown to have a D178N mutation in the PrP gene associated with fatal familial insomnia (FFI). The remaining 9 patients were diagnosed with sporadic Creutzfeldt-Jakob disease (sCJD); diagnosis was confirmed in 6 of them.8
Women accounted for 71.4% of patients in our series. Age at onset ranged from 27 to 77 years (median=54 years). Patients with a family history were younger (median=44.5 years). Survival time in the patient with vCJD was 6 months. The median survival time in patients with sCJD was 9.5 months. Patients with familial prion disease showed a mean survival time of 20 months.
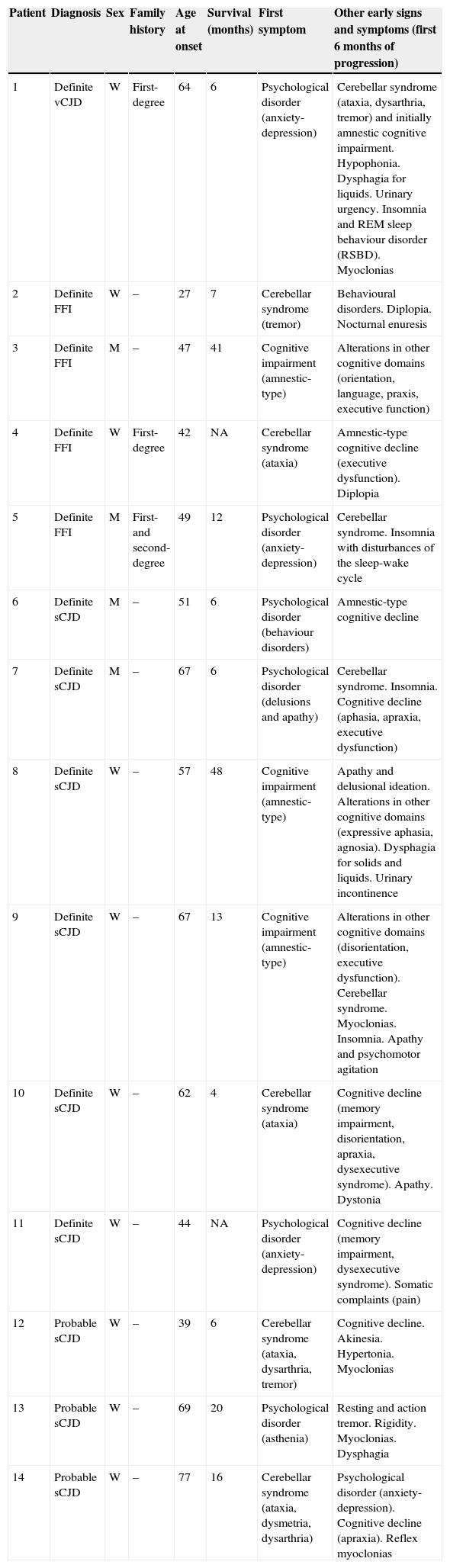
In 42.85% of the patients, mood and behaviour changes were the first clinical symptoms of the disease, which is characterised in most cases by anxiety-depressive disorder with a tendency to apathy in addition to delusional ideation. In 35.71%, the disease appeared as cerebellar syndrome with ataxic gait and postural and kinetic tremor. The initial reason for consultation in the 3 remaining patients was rapidly progressive isolated cognitive decline, and memory problems were the main complaint in these cases. However, a thorough neuropsychological evaluation revealed more extensive impairment affecting other cognitive domains that included orientation, language, praxis, and executive function. We emphasise that although patients reported one of the above symptoms as the initial symptom, the first neurological evaluation revealed multiple symptoms in every case. Other frequent signs and symptoms during the first 6 months of progression were myoclonias (in 5 out of 14 patients), urinary incontinence, dysphagia, and muscular rigidity. Patient characteristics are shown in Table 1.
Clinical and epidemiological characteristics
| Patient | Diagnosis | Sex | Family history | Age at onset | Survival (months) | First symptom | Other early signs and symptoms (first 6 months of progression) |
|---|---|---|---|---|---|---|---|
| 1 | Definite vCJD | W | First-degree | 64 | 6 | Psychological disorder (anxiety-depression) | Cerebellar syndrome (ataxia, dysarthria, tremor) and initially amnestic cognitive impairment. Hypophonia. Dysphagia for liquids. Urinary urgency. Insomnia and REM sleep behaviour disorder (RSBD). Myoclonias |
| 2 | Definite FFI | W | – | 27 | 7 | Cerebellar syndrome (tremor) | Behavioural disorders. Diplopia. Nocturnal enuresis |
| 3 | Definite FFI | M | – | 47 | 41 | Cognitive impairment (amnestic-type) | Alterations in other cognitive domains (orientation, language, praxis, executive function) |
| 4 | Definite FFI | W | First-degree | 42 | NA | Cerebellar syndrome (ataxia) | Amnestic-type cognitive decline (executive dysfunction). Diplopia |
| 5 | Definite FFI | M | First- and second-degree | 49 | 12 | Psychological disorder (anxiety-depression) | Cerebellar syndrome. Insomnia with disturbances of the sleep-wake cycle |
| 6 | Definite sCJD | M | – | 51 | 6 | Psychological disorder (behaviour disorders) | Amnestic-type cognitive decline |
| 7 | Definite sCJD | M | – | 67 | 6 | Psychological disorder (delusions and apathy) | Cerebellar syndrome. Insomnia. Cognitive decline (aphasia, apraxia, executive dysfunction) |
| 8 | Definite sCJD | W | – | 57 | 48 | Cognitive impairment (amnestic-type) | Apathy and delusional ideation. Alterations in other cognitive domains (expressive aphasia, agnosia). Dysphagia for solids and liquids. Urinary incontinence |
| 9 | Definite sCJD | W | – | 67 | 13 | Cognitive impairment (amnestic-type) | Alterations in other cognitive domains (disorientation, executive dysfunction). Cerebellar syndrome. Myoclonias. Insomnia. Apathy and psychomotor agitation |
| 10 | Definite sCJD | W | – | 62 | 4 | Cerebellar syndrome (ataxia) | Cognitive decline (memory impairment, disorientation, apraxia, dysexecutive syndrome). Apathy. Dystonia |
| 11 | Definite sCJD | W | – | 44 | NA | Psychological disorder (anxiety-depression) | Cognitive decline (memory impairment, dysexecutive syndrome). Somatic complaints (pain) |
| 12 | Probable sCJD | W | – | 39 | 6 | Cerebellar syndrome (ataxia, dysarthria, tremor) | Cognitive decline. Akinesia. Hypertonia. Myoclonias |
| 13 | Probable sCJD | W | – | 69 | 20 | Psychological disorder (asthenia) | Resting and action tremor. Rigidity. Myoclonias. Dysphagia |
| 14 | Probable sCJD | W | – | 77 | 16 | Cerebellar syndrome (ataxia, dysmetria, dysarthria) | Psychological disorder (anxiety-depression). Cognitive decline (apraxia). Reflex myoclonias |
We screened for 14-3-3 protein in the CSF11 in 11 patients: results were positive in 7 cases (2 patients with FFI and 5 with sCJD) after a mean progression time of 5.14 months. Results were negative in the remaining 4 patients, and mean progression times at evaluation were similar (5.5 months, SD 2.51).
EEG was performed in 12 patients. Results were normal in 1 patient with FFI (at 8 months of progression) and non-specific alterations were found in 9 patients (1 patient with vCJD, 2 with FFI, and 6 with sCJD; mean progression time=4.89 years, SD 2.71). These abnormalities consisted of non-specific background activity featuring semi-continuous slow polymorphic theta-delta waves that were either diffuse or localised. Only 2 patients with a diagnosis of sCJD displayed a pattern typical of prion disease, i.e. bilateral periodic diffuse discharges, predominantly in the anterior area, with a short-interval triphasic pattern (1Hz). These patients were studied with EEG at 3 and 4 months of progression, respectively.
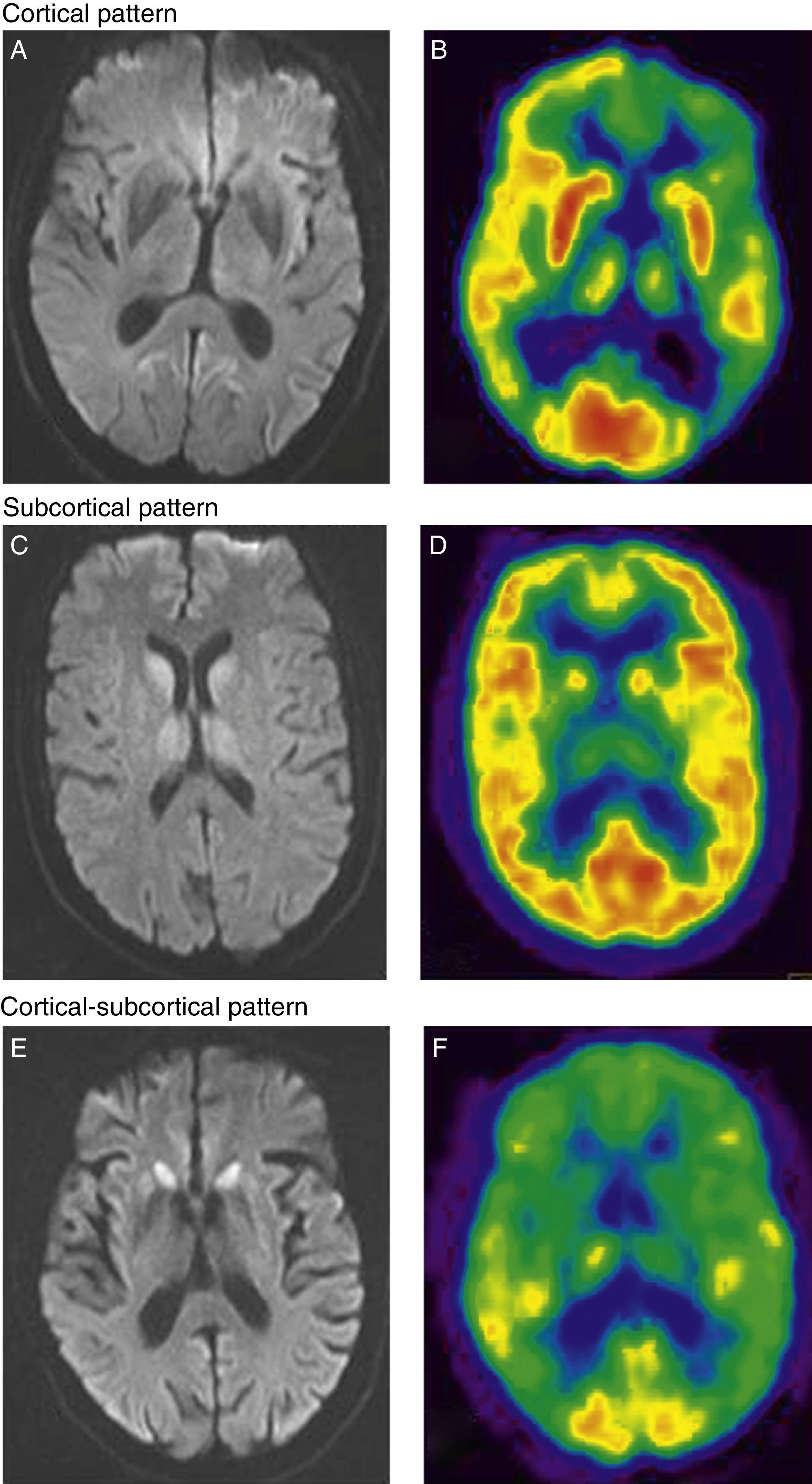
Regarding neuroimaging results, the 13 patients who underwent MRI and/or FDG-PET studies displayed anomalies suggestive of a prion disease (Fig. 1). In patients who underwent MRI and FDG-PET testing, both studies were performed at the same point in disease progression. Eleven patients underwent brain MRI studies with the sequences recommended for diagnosing CJD (FLAIR and DWI). Patients with the vCJD form displayed isolated bilateral thalamic hyperintensity,10 although the pulvinar sign typical of the disease was not observed in any of them.12 MRI studies were also performed on 2 patients with FFI who were not relatives. One image showed a subcortical pattern, and the other showed cortical hyperintensity, especially in the limbic and occipital regions.11
 Cortical hyperintensity in frontal-parietal-occipital regions of the left hemisphere in DWI sequences. (B) FDG-PET shows hypometabolism in the cortical-parietal, posterior cingulate, and dorsolateral frontal regions of the left hemisphere, as well as in the left basal ganglia and both thalamic hemispheres. Subcortical pattern. (C) Subcortical hyperintensity (basal ganglia and thalamus bilaterally) in DWI sequences. (D) FDG-PET scan shows hypometabolism in the left temporal cortical and subcortical regions (bilateral in thalamus). Cortical-subcortical pattern. (E) Bilateral symmetrical hyperintensity in frontal cortical and parietal regions; subcortical hyperintensity (caudate nucleus and putamen) in DWI sequences. (F) FDG-PET shows hypometabolism in both cortical areas (anterior association area and cingulate cortex) and subcortical areas (basal ganglia).")
Cortical pattern. (A) Cortical hyperintensity in frontal-parietal-occipital regions of the left hemisphere in DWI sequences. (B) FDG-PET shows hypometabolism in the cortical-parietal, posterior cingulate, and dorsolateral frontal regions of the left hemisphere, as well as in the left basal ganglia and both thalamic hemispheres. Subcortical pattern. (C) Subcortical hyperintensity (basal ganglia and thalamus bilaterally) in DWI sequences. (D) FDG-PET scan shows hypometabolism in the left temporal cortical and subcortical regions (bilateral in thalamus). Cortical-subcortical pattern. (E) Bilateral symmetrical hyperintensity in frontal cortical and parietal regions; subcortical hyperintensity (caudate nucleus and putamen) in DWI sequences. (F) FDG-PET shows hypometabolism in both cortical areas (anterior association area and cingulate cortex) and subcortical areas (basal ganglia).
In patients with sCJD, the predominant pattern was cortical-subcortical (4 of the 8 patients with MRI studies). Two patients showed an exclusively cortical pattern, and another patient had a subcortical pattern (Fig. 1).
On the other hand, 10 patients underwent FDG-PET testing and another was studied with 99mTc-HMPAO SPECT. In the patient with vCJD, the FDG-PET study showed cortical hypometabolism located predominantly in the left frontal and parietal regions, in addition to the bilateral thalamic hypometabolism that had been revealed by the MRI study.10 Two out of the 3 patients with FFI examined with FDG-PET imaging displayed an exclusively cortical pattern, and one showed a larger hypometabolic area that also extended to cortical regions.13 One patient with sCJD was studied with 99mTc-HMPAO SPECT, and another 7 underwent FDG-PET. This group was characterised by a cortical-subcortical pattern (6 out of 8 patients). The remaining 2 patients showed hypometabolism in the subcortical region.
Comparison of these 2 imaging techniques revealed FDG-PET to be more sensitive than MRI in 7 patients: hyperintense areas shown in MRI studies could also be seen in FDG-PET studies, whereas the opposite was not true. This technique was useful for identifying additional regions of the brain with altered metabolism. In 2 patients, both imaging techniques yielded similar results. Only in one case did MRI studies detect a greater number of affected areas than FDG-PET studies.
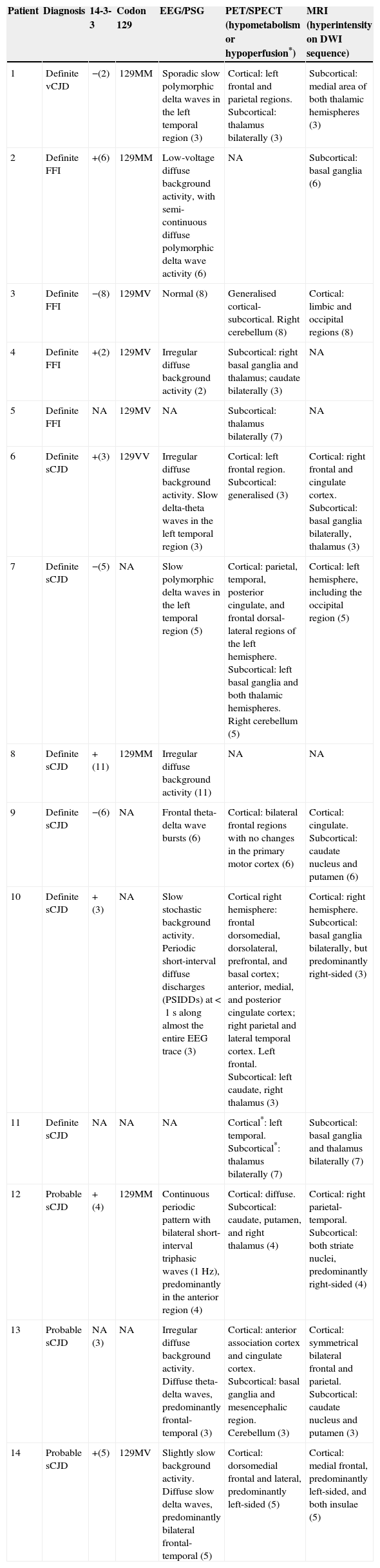
Genetic studies of PRNP were performed in 9 patients, and post-mortem histological studies in 7. Table 2 summarises the most significant findings from diagnostic tests.
Additional tests
| Patient | Diagnosis | 14-3-3 | Codon 129 | EEG/PSG | PET/SPECT (hypometabolism or hypoperfusion*) | MRI (hyperintensity on DWI sequence) |
|---|---|---|---|---|---|---|
| 1 | Definite vCJD | −(2) | 129MM | Sporadic slow polymorphic delta waves in the left temporal region (3) | Cortical: left frontal and parietal regions. Subcortical: thalamus bilaterally (3) | Subcortical: medial area of both thalamic hemispheres (3) |
| 2 | Definite FFI | +(6) | 129MM | Low-voltage diffuse background activity, with semi-continuous diffuse polymorphic delta wave activity (6) | NA | Subcortical: basal ganglia (6) |
| 3 | Definite FFI | −(8) | 129MV | Normal (8) | Generalised cortical-subcortical. Right cerebellum (8) | Cortical: limbic and occipital regions (8) |
| 4 | Definite FFI | +(2) | 129MV | Irregular diffuse background activity (2) | Subcortical: right basal ganglia and thalamus; caudate bilaterally (3) | NA |
| 5 | Definite FFI | NA | 129MV | NA | Subcortical: thalamus bilaterally (7) | NA |
| 6 | Definite sCJD | +(3) | 129VV | Irregular diffuse background activity. Slow delta-theta waves in the left temporal region (3) | Cortical: left frontal region. Subcortical: generalised (3) | Cortical: right frontal and cingulate cortex. Subcortical: basal ganglia bilaterally, thalamus (3) |
| 7 | Definite sCJD | −(5) | NA | Slow polymorphic delta waves in the left temporal region (5) | Cortical: parietal, temporal, posterior cingulate, and frontal dorsal-lateral regions of the left hemisphere. Subcortical: left basal ganglia and both thalamic hemispheres. Right cerebellum (5) | Cortical: left hemisphere, including the occipital region (5) |
| 8 | Definite sCJD | +(11) | 129MM | Irregular diffuse background activity (11) | NA | NA |
| 9 | Definite sCJD | −(6) | NA | Frontal theta-delta wave bursts (6) | Cortical: bilateral frontal regions with no changes in the primary motor cortex (6) | Cortical: cingulate. Subcortical: caudate nucleus and putamen (6) |
| 10 | Definite sCJD | + (3) | NA | Slow stochastic background activity. Periodic short-interval diffuse discharges (PSIDDs) at <1s along almost the entire EEG trace (3) | Cortical right hemisphere: frontal dorsomedial, dorsolateral, prefrontal, and basal cortex; anterior, medial, and posterior cingulate cortex; right parietal and lateral temporal cortex. Left frontal. Subcortical: left caudate, right thalamus (3) | Cortical: right hemisphere. Subcortical: basal ganglia bilaterally, but predominantly right-sided (3) |
| 11 | Definite sCJD | NA | NA | NA | Cortical*: left temporal. Subcortical*: thalamus bilaterally (7) | Subcortical: basal ganglia and thalamus bilaterally (7) |
| 12 | Probable sCJD | + (4) | 129MM | Continuous periodic pattern with bilateral short-interval triphasic waves (1Hz), predominantly in the anterior region (4) | Cortical: diffuse. Subcortical: caudate, putamen, and right thalamus (4) | Cortical: right parietal-temporal. Subcortical: both striate nuclei, predominantly right-sided (4) |
| 13 | Probable sCJD | NA (3) | NA | Irregular diffuse background activity. Diffuse theta-delta waves, predominantly frontal-temporal (3) | Cortical: anterior association cortex and cingulate cortex. Subcortical: basal ganglia and mesencephalic region. Cerebellum (3) | Cortical: symmetrical bilateral frontal and parietal. Subcortical: caudate nucleus and putamen (3) |
| 14 | Probable sCJD | +(5) | 129MV | Slightly slow background activity. Diffuse slow delta waves, predominantly bilateral frontal-temporal (5) | Cortical: dorsomedial frontal and lateral, predominantly left-sided (5) | Cortical: medial frontal, predominantly left-sided, and both insulae (5) |
Months of disease progression at the time each additional test was conducted shown in brackets.
NA: not available.
* Patient no. 11 showed cortical and subcortical hypoperfusion in SPECT.
This article presents a description of 14 patients from different regions of Spain (Navarre, the Basque Country, Madrid, Andalusia, Valencia, Castile-Leon, and La Rioja), and with different forms of prion diseases, who were examined in our department between 1999 and 2012.
In our series, cases of sCJD were more frequent than FFI or vCJD, although these last 2 forms of prion disease account for a large percentage of our series due to a reference standard bias. Only one patient experienced onset of the disease when younger than 50 years, the typical age of onset for sCJD.14 Regarding the familial form of CJD, all patients were carriers of the D178N mutation, which an earlier study has described as a frequent mutation among families with prion disease in the Basque Country.15 In the patient with vCJD, clinical symptoms appeared later than normal7 and survival time was shorter.
The most frequent initial symptoms in our patient series were mood and behaviour disorders, while other studies have listed cognitive decline5 or cerebellar syndrome.5,16 Initial symptoms vary by patient series, which can therefore result in late diagnosis.5 Nevertheless, all authors agree that a large percentage of patients simultaneously present 3 types of symptoms: cognitive decline, anxiety-depressive disorders, and cerebellar syndrome. Based on the literature and our own data, we believe that the presence of this triad of symptoms is highly indicative of some type of prion disease.
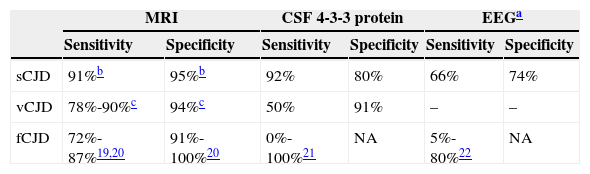
Additional tests are very useful for diagnosing these diseases. Presence of 14-3-3 protein in CSF is a marker of neuronal damage that indicates prion disease.17 Previous studies have shown that detecting 14-3-3 protein in the CSF is sensitive and specific for a diagnosis of sCJD. In 2009, this finding was included as one of the diagnostic criteria for the disease.8 In a recent review including patients with a probable or definite diagnosis of sCJD, Muayqil et al.18 estimate that 14-3-3 protein detection has a sensitivity of 92% and a specificity of 80% (Table 3). Presence of 14-3-3 protein and typical abnormalities in the EEG (periodic sharp wave complexes)8 have a strong positive predictive value for CJD,23 whereas other diagnoses should be considered in the absence of one or both findings.23 However, these 2 tests may also yield false positive results. The study by Tschampa et al. reports a series of patients diagnosed with prion disease whose post-mortem study revealed findings consistent with Alzheimer disease or Lewy body dementia.24 Two of our patients with a definite diagnosis of sCJD did not show typical abnormalities in the EEG, and 14-3-3 protein tests yielded negative results. According to these data, diagnosis of prion disease should not be ruled out in cases in which results from EEG and 14-3-3 protein tests are negative if clinical suspicion is strong and other possible causes have already been ruled out. The diagnostic value of CSF 14-3-3 protein for familial CJD is lower.23 In our series, however, 14-3-3 protein yielded positive results in 2 of the 3 patients who underwent testing.
Sensitivity and specificity of the additional tests
| MRI | CSF 4-3-3 protein | EEGa | ||||
|---|---|---|---|---|---|---|
| Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity | |
| sCJD | 91%b | 95%b | 92% | 80% | 66% | 74% |
| vCJD | 78%-90%c | 94%c | 50% | 91% | – | – |
| fCJD | 72%-87%19,20 | 91%-100%20 | 0%-100%21 | NA | 5%-80%22 | NA |
sCJD: sporadic Creutzfeldt-Jakob disease; fCJD: familial Creutzfeldt-Jakob disease; vCJD: variant Creutzfeldt-Jakob disease; NA: not available.
In the past few years, neuroimaging studies have become especially relevant in diagnosing prion diseases.25 Characteristic brain MRI findings have been included among the diagnostic criteria for vCJD7 and sCJD.8 The presence of the pulvinar sign in brain MRI is a finding supporting vCJD diagnosis12 and has a sensitivity ranging from 78% to 90% (Table 3). It consists of bilateral hyperintensity of the pulvinar area compared to the signal intensity of the cortex and anterior putamen. Correctly identifying this radiological finding is important since patients with sCJD may present a false-positive pulvinar sign in which hyperintensity in the pulvinar nuclei is less marked than in the putamen and caudate.9 The combination of FLAIR and DWI sequences in brain MRI has a higher detection capacity for sCJD than 14-3-3 protein and EEG,26,27 with a diagnostic sensitivity of 91% and a specificity of 95%.28 We have imaging study results from 7 of the 9 patients with sCJD in our series. All of them showed typical findings (signal alterations in both the caudate nucleus and putamen, or in at least 2 cortical regions).8 These 7 patients also underwent a FDG-PET scan, which revealed cortical-subcortical hypometabolism. Familial forms of prion disease are not currently known to have specific MRI patterns, although certain non-specific changes have been described (brain ventricle dilation, cerebellar and cerebral cortex atrophy). Brain metabolism studies completed in 3 patients with familial forms of prion disease showed extensive hypometabolic areas in the thalamus. These findings are consistent with the literature.29,30 According to these data, cortical-subcortical hypometabolism in FDG-PET may be more typical of the sporadic forms of the disease, while familial forms are more likely to present thalamic hypometabolism.
The data from our patient series support the importance of neuroimaging techniques. MRI and/or FDG-PET tests were decisive for achieving diagnosis in all patients who underwent neuroimaging studies (13), and in 5 patients with a definitive diagnosis, these tests were the only ones to yield positive results.
In conclusion, the combination of psychological, cognitive, and cerebellar symptoms is strongly indicative of prion disease. Regarding additional tests, neuroimaging studies (MRI and/or FDG-PET) offer a high degree of sensitivity and are therefore helpful in the diagnosis of prion diseases. In our series, FDG-PET scans were useful for identifying additional brain regions with metabolic alterations, and they were shown to have a greater diagnostic ability than MRI. This has also been suggested by recently published articles.31,32
Therefore, if prion disease is clinically suspected, it is essential to perform a brain MRI (DWI and FLAIR sequences) and/or a FDG-PET scan. Although FDG-PET studies are not currently included among the diagnostic criteria for prion diseases, our data suggest that this test may play a key role in future diagnoses.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Ortega-Cubero S, Pagola I, Luquin MR, Viteri C, Pastor P, Gállego Pérez-Larraya J, et al. Descripción de una serie de pacientes con diagnóstico de enfermedad priónica. Neurología. 2015;30:144–152.
This study was presented orally at the 64th Annual Meeting of the Spanish Society of Neurology in November 2012.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas