



We present the case of a 63-year-old woman who had been diagnosed 4 years earlier with multiple sclerosis (MS) according to McDonald criteria due to a clinically isolated demyelinating syndrome and MRI findings of multiple cerebral and brainstem lesions compatible with demyelinating aetiology.
She returned to the clinic due to experiencing poor balance and vision changes consisting of diplopia and loss of voluntary gaze; symptoms had begun 15 days before.
Neurological examination revealed total horizontal ophthalmoplegia (lack of saccades and gaze following; no eye movements with doll's head manoeuvre). Convergence was preserved and there were no limits along the vertical axis and no associated pupil changes; nystagmus appeared with upgaze. We observed no facial weakness or effects on other cranial nerves, and all other findings were normal.
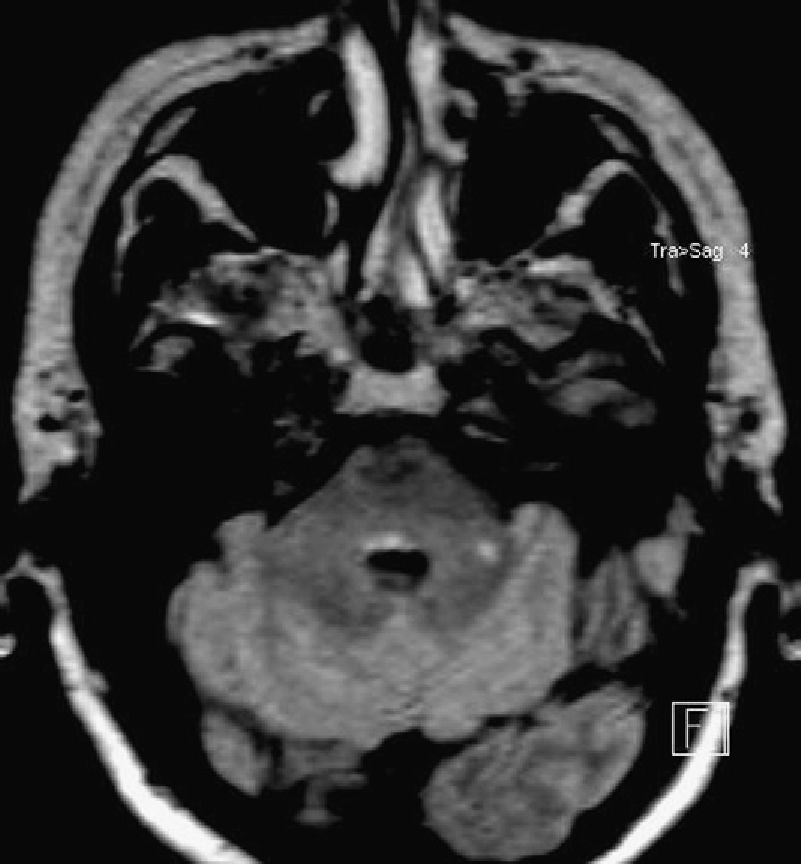
Brain MRI using long TR sequences showed extensive hyperintensities merging together along the calloseptal interface and periventricular deep white matter; these features were already known. The scan also showed a small hyperintensity surrounding the fourth ventricle in the posterior pontine region that had not appeared in previous studies (Figs. 1 and 2).


CSF study revealed oligoclonal bands of IgG, but they were absent in serum; there was also an increase in intrathecal IgG production. Additional tests (full blood count, biochemistry test, autoimmunity test, serology test) all yielded normal results.
The patient was treated with IV bolus of methylprednisolone dosed at 1g/24h over 5 days. Ocular palsy was observed to improve slightly; bilateral internuclear ophthalmoplegia and partial, asymmetrical recovery of abduction were also apparent. By the last check-up, performed 6 months after symptom onset, recovery was complete.
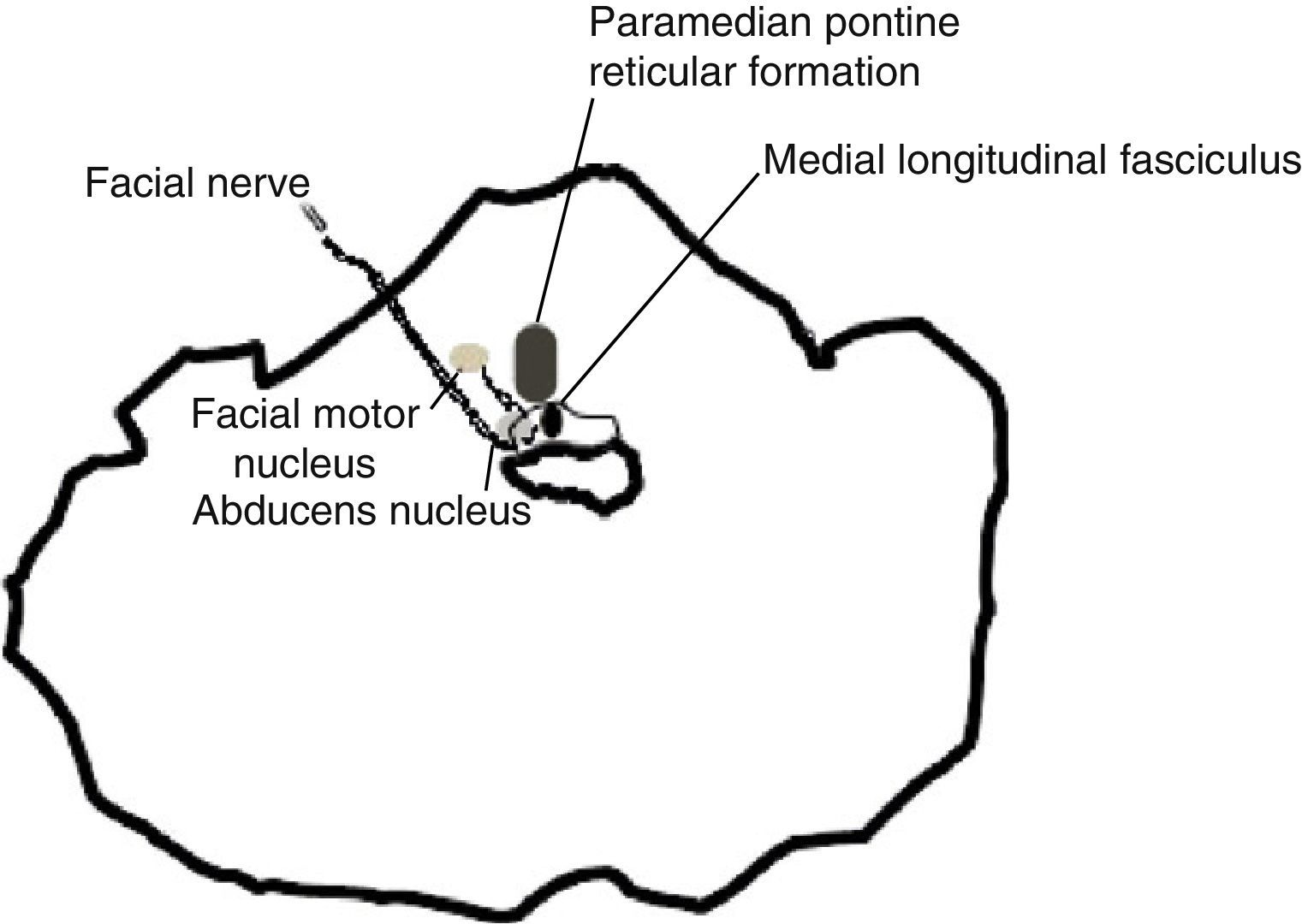
Oculomotor impairment is a very frequent finding in the course of MS. Of all the different types, internuclear ophthalmoplegia, which results from lesion to the medial longitudinal fasciculus, is considered a sign of the brainstem impairment typical of the disease. Extension of the lesion, which by its proximity will affect the abducens nucleus and/or adjacent pontine reticular formation, will result in horizontal ophthalmoplegia in which only the eye contralateral to the lesion performs adduction; this has been called ‘one and a half syndrome’.1
Far more uncommon is the appearance of complete horizontal ophthalmoplegia as the cause of a demyelinating lesion like the one described here. Very few cases have been published in the medical literature. This syndrome was recently named ‘1+1 syndrome’.2
This case displays bilateral impairment of the abducens nucleus and adjacent pontine reticular formation. It has also been described as the result of damage to the fibres of the fourth pair at the emergence of both nuclei with impairment of the medial longitudinal fasciculus.3
Impairment of the abducens nucleus tends to be accompanied by impairment of the facial nerve that surrounds it before continuing on to emerge laterally in the pons.4
In our case, absence of facial palsy indicates that the former mechanism is more likely, as can be deduced by the brain MRI scan. One case series on ophthalmoplegia in which researchers correlated clinical findings with cranial MRI findings includes a case of complete horizontal gaze palsy with a discrete lesion restricted to the midline of the dorsal pons. There was no apparent compromise of the abducens nucleus or the paramedian pontine reticular formation, as in the case we present. In our case, independent from impairment of the medial longitudinal fasciculus, the nucleus raphe interpositus ventral to that structure may be responsible for gaze paralysis.5
Please cite this article as: Vidal de Francisco D, Argente Alcaraz J, Espinosa Rosso R. Oftalmoplejía internuclear horizontal bilateral completa; síntoma de presentación de un brote de esclerosis múltiple. Neurología. 2014;29:252–253.
