



Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome is a rare multisystemic disease of paraneoplastic origin, which represents a true clinical challenge. It is caused by a plasma cell disease and its symptoms are numerous and varied. The acronym refers to the combination of the most frequently manifesting signs and symptoms: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.1 However, some of these symptoms are absent in some cases, and are not necessary for establishing diagnosis. It has been suggested that overproduction of vascular endothelial growth factor (VEGF), secreted by neoplastic plasma cells, may be responsible for most symptoms.2 The diagnostic criteria are:
- •
Major criteria (both mandatory): polyneuropathy (mainly demyelinating) and monoclonal proliferation of plasma cells.
- •
Major criteria (at least one must be present): Castleman disease, sclerotic bone lesions, and increased VEGF levels.
- •
Minor criteria (at least one must be present): organomegaly (splenomegaly, hepatomegaly, adenopathies); extravascular volume overload (oedemas, pleural oedema, ascites); endocrinopathies (adrenal, thyroid, pituitary, pancreatic); skin changes (hyperpigmentation, hypertrichosis, acrocyanosis, white nails); papilloedema; and thrombocytosis/polycythaemia.
We present a case of POEMS syndrome in a 71-year-old woman who was transferred to the haematology department for a polycythaemia study after a stroke. Her personal history included arterial hypertension, untreated C virus–related chronic liver disease, and Paget disease of bone; she was under follow-up by the neurology department due to a 3-year history of mixed progressive polyneuropathy, with no response to several lines of treatment. Blood analysis showed a haemoglobin level of 18.6g/dL and a haematocrit level of 58%. The rest of the examination yielded normal results. Erythropoietin levels were normal and the patient was negative for the v617f mutation of the JAK2 gene. A screening test for acquired and inherited thrombophilia obtained negative results.
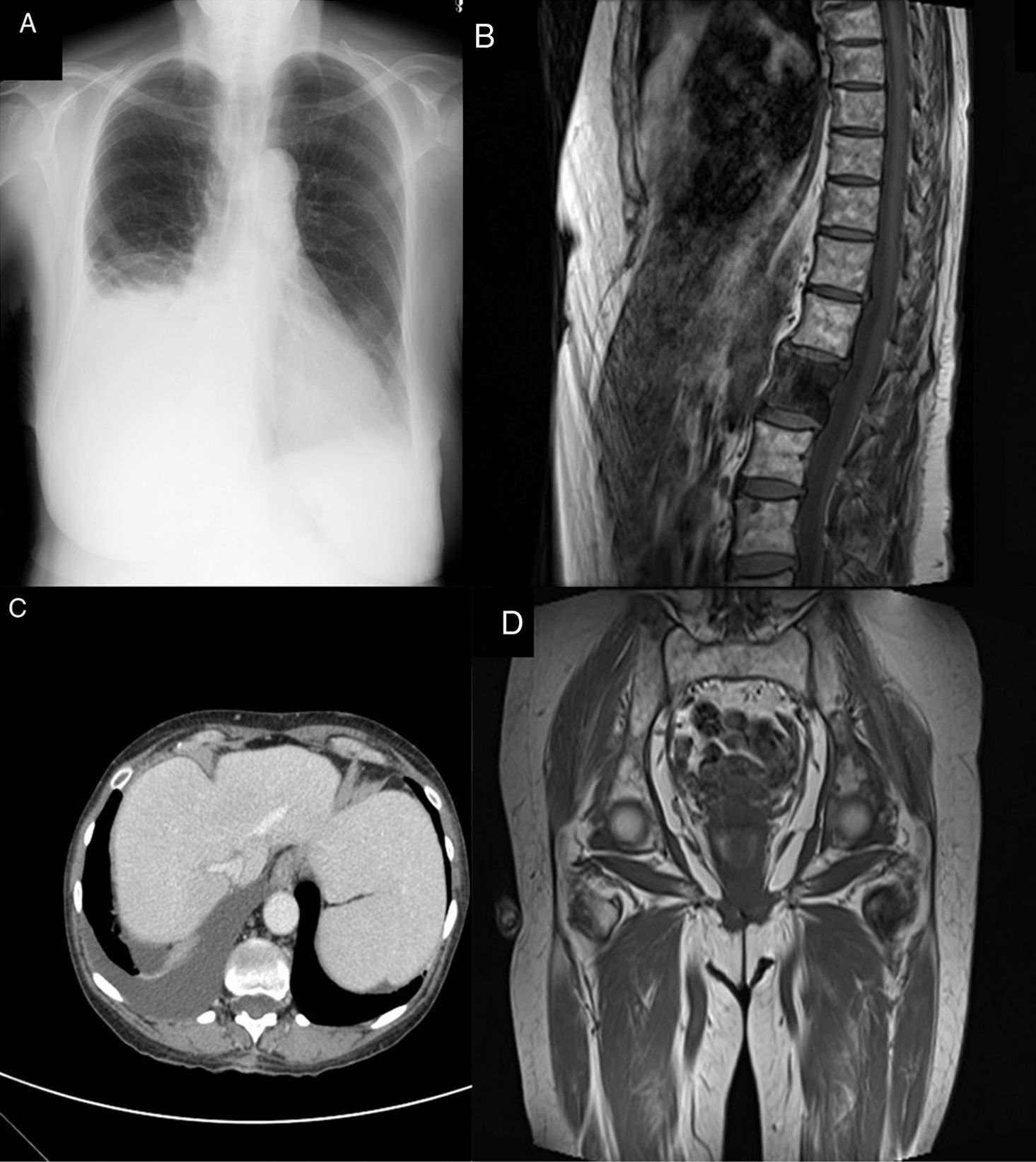
Further testing revealed low levels of monoclonal IgA-lambda protein, which was not detectable in the urine. During follow-up, the patient attended the emergency department due to dyspnoea with mild exertion, and a bilateral pleural effusion with transudate features (cardiac and infectious causes were ruled out) (Fig. 1A). A high-resolution thoracic CT scan revealed sclerotic bone lesions in the right fifth rib and the L1 vertebra (Fig. 1B). A magnetic resonance imaging scan and full CT scan showed multiple osteosclerotic lesions, hepatosplenomegaly, and bilateral pleural effusion with minimal free abdominal fluid (Fig. 1C). As POEMS syndrome was suspected, we requested a VEGF determination (>1000pg/mL; normal values, <128pg/mL) and biopsy of the rib lesion (not conclusive). Bone marrow biopsy revealed no pathological infiltration. The patient met 2 mandatory criteria, 2 major criteria, and several minor criteria; therefore, diagnosis of POEMS syndrome was established.
 Bilateral pleural effusion, more pronounced in the right hemithorax. (B) Sclerotic lesion affecting vertebra L1. (C) Hepatosplenomegaly and right pleural effusion. (D) Sclerotic lesion in the left minor trochanter.")
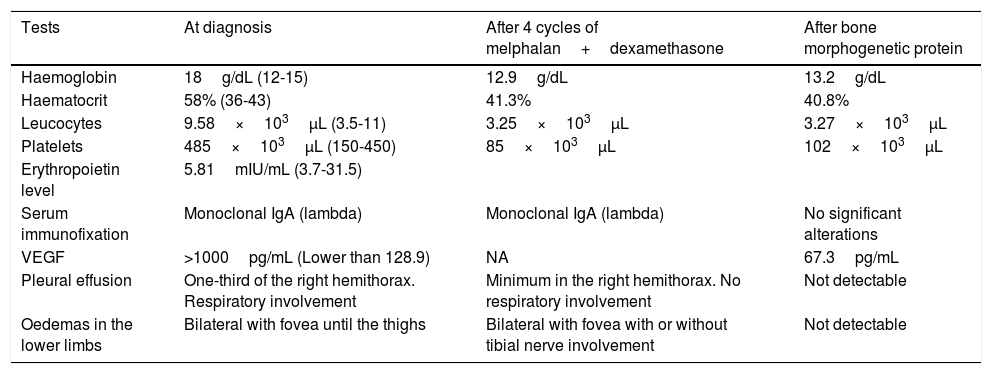
Considering the patient's age and comorbidities, an autologous stem cell transplantation was ruled out2 and treatment with melphalan+dexamethasone was started. The patient's condition improved slightly, with decreased pleural effusion and reduced oedema of the lower limbs; repeated hospitalisation was not required. As polyneuropathy did not improve after 4 cycles of treatment, treatment was started with bortezomib. After 2 cycles, the paraprotein became undetectable and pleural effusion and oedema in the lower limbs disappeared. Haemoglobin and VEGF levels normalised and no new sclerotic lesions were detected on the follow-up magnetic resonance imaging scan. Eight months after finishing treatment, the patient remained stable and neuropathy slightly improved; he was able to start treatment for hepatitis C virus (Table 1).
Clinical and laboratory data during diagnosis and treatment.
| Tests | At diagnosis | After 4 cycles of melphalan+dexamethasone | After bone morphogenetic protein |
|---|---|---|---|
| Haemoglobin | 18g/dL (12-15) | 12.9g/dL | 13.2g/dL |
| Haematocrit | 58% (36-43) | 41.3% | 40.8% |
| Leucocytes | 9.58×103μL (3.5-11) | 3.25×103μL | 3.27×103μL |
| Platelets | 485×103μL (150-450) | 85×103μL | 102×103μL |
| Erythropoietin level | 5.81mIU/mL (3.7-31.5) | ||
| Serum immunofixation | Monoclonal IgA (lambda) | Monoclonal IgA (lambda) | No significant alterations |
| VEGF | >1000pg/mL (Lower than 128.9) | NA | 67.3pg/mL |
| Pleural effusion | One-third of the right hemithorax. Respiratory involvement | Minimum in the right hemithorax. No respiratory involvement | Not detectable |
| Oedemas in the lower limbs | Bilateral with fovea until the thighs | Bilateral with fovea with or without tibial nerve involvement | Not detectable |
POEMS syndrome is an infrequent entity which falls within the group of plasma cell dyscrasias. Given the rareness, variability, and complexity of forms of onset, diagnosis of POEMS syndrome may be delayed by a median of 13-18 months.3 Therefore, initial clinical suspicion, based on a good history taking and physical examination, is essential to identify which laboratory tests and radiological studies may ultimately enable us to establish a definitive diagnosis.
In our case, diagnosis was established 9 months after the initial visit to the haematology department, 3 years after symptom onset (neuropathy), and 5 years after the non-specific bone lesions were observed in a bone scintigraphy (attributed to probable Paget disease of the bone).
Diagnostic delay may entail a decrease in the subsequent response to treatments, increasing sequelae and complications4; therefore, one of the main challenges in the context of this disease is to minimise time to diagnosis. Neuropathy is on many occasions the first manifestation, and becomes less reversible as disease progression time increases.
It is also important to highlight that some symptoms that are not included in the POEMS acronym may present prematurely and must be recognised, since they may be the first sign supporting suspicion of the disease. These include papilloedema, presence of oedemas (associated with an increased capillary permeability due to VEGF), sclerotic lesions, thrombocytosis/polycythaemia, or stroke (5-year risk of 13.4%),5 among others.
In the present case, the detection of a monoclonal paraprotein associated with presence of a mixed polyneuropathy led us to focus our diagnostic efforts by broadening our approach to screen for some of the other diagnostic criteria (VEGF levels, sclerotic bone lesions, etc.).
Evidence regarding treatment is limited and there are no guidelines supported by large prospective studies. There are a number of different therapeutic options, which usually improve some clinical manifestations, although polyneuropathy often responds more slowly.6 In patients younger than 65, autologous stem cell transplantation is the proposed first-line treatment.7 In patients older than 65 with associated comorbidities, the recommendations are to use chemotherapy regimens similar to those for multiple myeloma, based on alkylating agents (melphalan) and steroids (dexamethasone). An increasing number of series report using such new immunomodulatory drugs as lenalidomide, and more recently such proteasome inhibitors as bortezomib, excluded until now due to the known risk of drug-induced peripheral neuropathy.8 The discovery of VEGF and its role in the aetiopathogenesis of the disease has enabled us to better understand the pathophysiology of the syndrome and to explain some of its clinical manifestations. However, the desired results were not obtained with targeted treatments which have not yet been demonstrated to be effective.6
Clinical suspicion is therefore essential for establishing an early diagnosis. This will provide patients with a specific treatment adapted to their situation, avoiding potential complications and decreasing sequelae, and especially neurological sequelae. Given the greatly variable forms of onset of the syndrome, clinicians should be aware of the multiple manifestations of the disease and perform a directed clinical, analytical, and radiological search.
Please cite this article as: Plaza C, Arquero T, García-Raso A, Llamas P. Diagnóstico de síndrome de POEMS tras neuropatía de larga evolución. Neurología. 2019;34:272–274.
