



El término “kernicterus” se aplicó inicialmente a la tinción amarilla de los ganglios basales en estudios necrópsicos, pero es un término impreciso y se habla más de encefalopatía por bilirrubina o de disfunción neurológica inducida por la bilirrubina. Clínicamente la toxicidad por hiperbilirrubinemia puede ser muy variable y en países desarrollados tiende a desaparecer.
Material y métodosRevisamos una serie de 7 pacientes con encefalopatía por bilirrubina y diferentes grados de compromiso neurológico, atendidos en los últimos 10 años en el Servicio. Solo falleció un paciente en período neonatal con hiperbilirrubinemia, sepsis y fallo multiorgánico.
ResultadosLas causas etiológicas de la hiperbilirrubinemia fueron muy variadas. Los 7 pacientes presentaron ictericia, clínica neonatal, la neuroimagen ya permitió demostrar las lesiones en núcleo pálido con hiperintensidad de T1. Todos los pacientes presentaron manifestaciones clínicas en período neonatal, y secuelas neurológicas más o menos graves en los 6 supervivientes que se intentan correlacionar con los demás parámetros bioquímicos, clínicos, de neuroimagen y neurofisiológicos.
ConclusionesHemos constatado un incremento de las observaciones de disfunción neurológica inducida por la bilirrubina y nos planteamos conocer las causas de esta situación. La mayor supervivencia de los grandes prematuros, el aumento de la población inmigrante y la posibilidad del diagnóstico por neuroimagen contribuyen a este incremento. Continua siendo un reto para el neonatólogo y el neuropediatra evitar su presentación y minimizar los efectos de la toxicidad por bilirrubina en período neonatal.
“Kernicterus” is a term currently used to describe bilirrubin induced brain injury in the neuro-pathological studies. This is a confusing term and nowadays we prefer bilirrubin encephalopathy or bilirrubin induced neurological dysfunction. The clinical signs vary and it is clearly decreasing in prevalence in developed countries.
Material and methodsWe review a series of 7 patients with bilirrubin encephalopathy and variable neurological manifestations, who were seen in the Neuropaediatric Department in the last 10 years. Only one patient died in the neonatal period with hyperbilirubinaemia, sepsis and multi-organ failure.
ResultsDiverse aetiological factors were related to hyperbilirubinaemia. All patients had clinical symptoms due to hyperbilirubinaemia. Neuroimaging during the neonatal period showed involvement of the nucleus pallidus, with hyperintensity in T1 in the brain MR scan as the most consistent finding. All the patients who survived developed neurological signs and we try to correlate them with biochemical, clinical, neuroimaging and neurophysiological parameters.
ConclusionsAn increase in the number of patients with bilirrubin encephalopathy has been observed over the last few years, and we attempt to find out the causes. The increased survival of the low birth weight newborns, the increase in the immigration population and the use of diagnostic neuroimaging contribute to this increase. It is a great challenge for the neonatologist and for neuropaediatricians to prevent its occurrence and to minimise the effects of bilirrubin encephalopathy.
El término kernicterus se aplicó inicialmente para denominar la pigmentación amarilla de los ganglios basales en muestras patológicas, de pacientes que habían fallecido con ictericia por eritroblastosis fetal1. Posteriormente se aplicó el término a la encefalopatía estática con tetraparesia distónica y defectos auditivos, producida por hiperbilirrubinemia en período neonatal1,2.
Otros autores preconizan emplear el término kernicterus para cualquier manifestación debida a la hiperbilirrubinemia neonatal con clinica tan distinta, desde una leve hipoacusia neurosensorial a otras más graves como retardo mental, sordera o coreoatetosis2.
En la actualidad se prefiere el término encefalopatía bilirrubínica o disfunción neurológica inducida por bilirrubina para explicar todas y cada una de las manifestaciones de la hiperbilirrubinemia neonatal no tratada o insuficientemente tratada.
La bilirrubina –derivada del catabolismo de la hemoglobina– es un pigmento sumamente tóxico para los sistemas biológicos, particularmente el sistema nervioso1,2. La entrada de la bilirrubina no conjugada al interior de la neurona obedece a procesos bioquímicos no del todo conocidos pero sí aceptados como probables. Brodersen3 y Wennberg4 han propuesto un modelo por el cual la bilirrubina puede adaptarse a la membrana celular de la neurona mediante un delicado equilibrio, en el que intervienen, entre otros, los siguientes factores: concentración sérica de albúmina, pH del medio y los cambios en la estructura interna de la molécula de bilirrubina. El pigmento puede entrar y salir del citoplasma neuronal según la estabilidad de dichos factores3,5. Se ha comprobado experimentalmente la entrada de bilirrubina unida a albúmina a través de la barrera hematoencefálica (BHE) dañada por el uso de agentes hipertónicos. En el interior de la neurona, la bilirrubina produce una disrupción del gradiente de protones e interfiere directamente en los procesos oxidativos intramitocondriales, finalmente conduce a la apoptosis y necrosis neuronal2–4.
La introducción de la resonancia magnética (RM) en la práctica clínica diaria de las unidades neonatales ha permitido visualizar las lesiones del kernicterus, que antes solamente se podían evidenciar mediante estudios anatomo-patológicos. Excelentes estudios recientemente publicados tratan el tema6,7. En la fase aguda, estos cambios probablemente traducen la reacción gemistocítica de los astrocitos. Una vez superada la fase aguda, si las imágenes de RM persisten alteradas, los hallazgos probablemente reflejan la densa gliosis fibrilar pobre en contenido celular que aparece en la fase final8.
En la actualidad y por varios motivos que posteriormente analizaremos hemos observado un incremento del número de pacientes con encefalopatía atribuida a la hiperbilirrubinemia y por ello creemos de interés exponer nuestra experiencia y la revisión de la literatura.
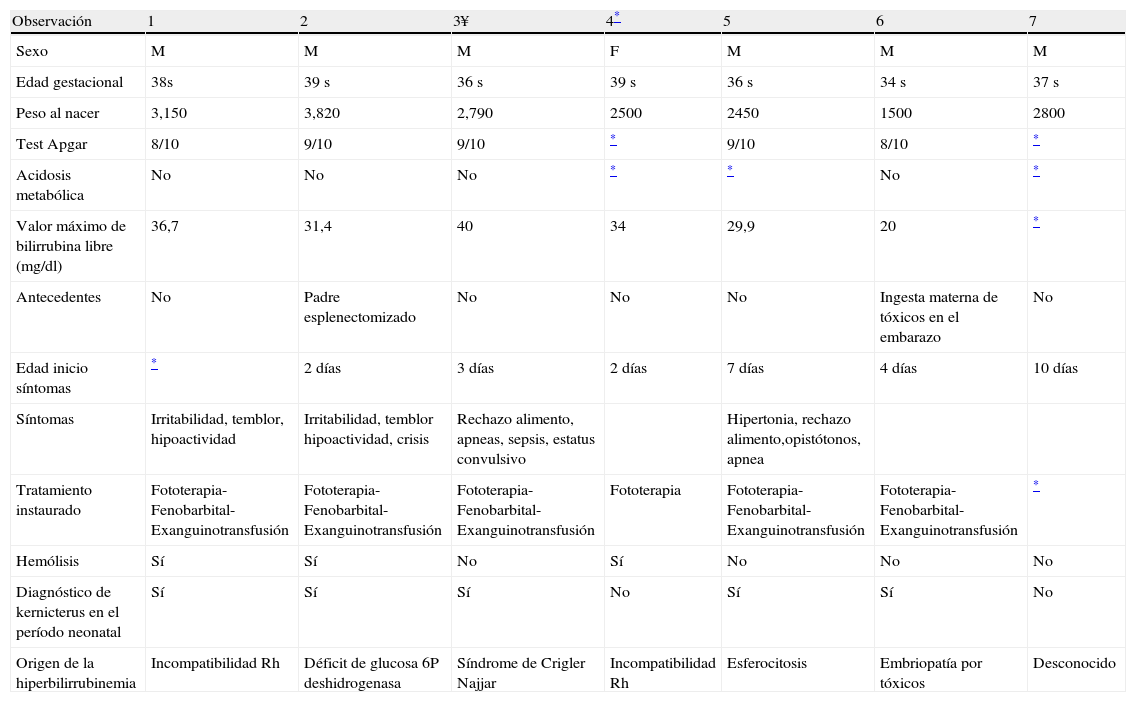
Pacientes y métodosSe revisaron las historias clínicas y los estudios de neuroimagen: RM y ecografías craneales transfontanelares de 7 pacientes atendidos en el Servicio de Neurología del Hospital, en el período 1999 y 2008; en los cuales fue posible determinar secuelas neurológicas atribuibles a la hiperbilirrubinemia neonatal (tabla 1). Seis pacientes son de sexo masculino y ninguno de los 7 pacientes había nacido en nuestro Hospital y dos de ellos fuera de España. Algunos fueron trasladados ya en período neonatal y otros vistos a posteriori. En el único paciente que falleció en nuestro centro se revisaron las muestras de tejido cerebral obtenidas en la necropsia. En la serie solamente dos pacientes no se diagnosticaran en período neonatal de encefalopatía por bilirrubina. En 3 casos se demostró hemólisis y el origen de la hiperbilirrubinemia fue muy diverso (incompatibilidad Rh dos casos y un caso de déficit de glucosa 6P deshidrogenasa, síndrome de Crigler Najjar, esferocitosis, embriopatía por tóxicos y otro de causa desconocida que había nacido fuera de España).
Resumen de los datos clínicos y bioquímicos de los pacientes afectos de kernicterus, (HSJD) 1998-2008
| Observación | 1 | 2 | 3¥ | 4* | 5 | 6 | 7 |
| Sexo | M | M | M | F | M | M | M |
| Edad gestacional | 38s | 39 s | 36 s | 39 s | 36 s | 34 s | 37 s |
| Peso al nacer | 3,150 | 3,820 | 2,790 | 2500 | 2450 | 1500 | 2800 |
| Test Apgar | 8/10 | 9/10 | 9/10 | * | 9/10 | 8/10 | * |
| Acidosis metabólica | No | No | No | * | * | No | * |
| Valor máximo de bilirrubina libre (mg/dl) | 36,7 | 31,4 | 40 | 34 | 29,9 | 20 | * |
| Antecedentes | No | Padre esplenectomizado | No | No | No | Ingesta materna de tóxicos en el embarazo | No |
| Edad inicio síntomas | * | 2 días | 3 días | 2 días | 7 días | 4 días | 10 días |
| Síntomas | Irritabilidad, temblor, hipoactividad | Irritabilidad, temblor hipoactividad, crisis | Rechazo alimento, apneas, sepsis, estatus convulsivo | Hipertonia, rechazo alimento,opistótonos, apnea | |||
| Tratamiento instaurado | Fototerapia-Fenobarbital-Exanguinotransfusión | Fototerapia- Fenobarbital-Exanguinotransfusión | Fototerapia- Fenobarbital-Exanguinotransfusión | Fototerapia | Fototerapia- Fenobarbital-Exanguinotransfusión | Fototerapia- Fenobarbital-Exanguinotransfusión | * |
| Hemólisis | Sí | Sí | No | Sí | No | No | No |
| Diagnóstico de kernicterus en el período neonatal | Sí | Sí | Sí | No | Sí | Sí | No |
| Origen de la hiperbilirrubinemia | Incompatibilidad Rh | Déficit de glucosa 6P deshidrogenasa | Síndrome de Crigler Najjar | Incompatibilidad Rh | Esferocitosis | Embriopatía por tóxicos | Desconocido |
En la tabla 1 se hallan en forma resumida los datos de los 7 pacientes. Solamente uno de ellos era pretérmino (34s) y todos los demás a término y con peso al nacimiento adecuado para la edad gestacional. Ninguno de los pacientes presentó problemas durante el parto, con valores normales en el test de Apgar. No se evidenció acidosis metabólica y destacar en dos casos historia de padre esplenectomizado en uno y de ingesta materna de drogas durante embarazo en otra observación. En todos los casos la clínica debutó en la primera semana de vida y muy particularmente en las primeras 72 horas de vida, con ictericia, irritabilidad, temblor, hipoactividad y movimientos anormales. La bilirrubina no conjugada máxima alcanzada por los pacientes se situó en un rango de 20-40mg/dl, entre los 2 y 10 días de vida. Todos los pacientes fueron tratados con fenobarbital, fototerapia intensiva y exanguinotransfusión con resultados dispares. Las cifras de bilirrubina se normalizaron entre los 6-10 días de vida.
Todos los pacientes estudiados presentaron alteraciones en el EEG de diversa naturaleza. Tres pacientes manifestaron crisis epilépticas en período neonatal, y uno de ellos falleció en la UCI neonatal con sepsis, estatus convulsivo, fallo multiorgánico y coma. Los demás presentan secuelas neurológicas consistentes en hipoacusia neurosensorial y tetraparesia mixta. El diagnóstico de encefalopatía por hiperbilirrubinemia fue realizado en el período neonatal en 5 observaciones en los 18 primeros días de vida y a posteriori durante el seguimiento en las demás.
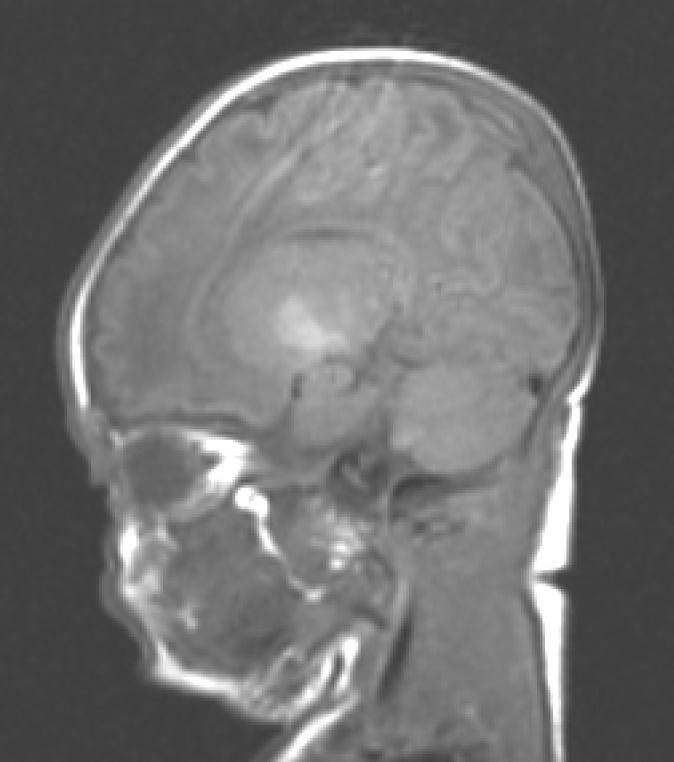
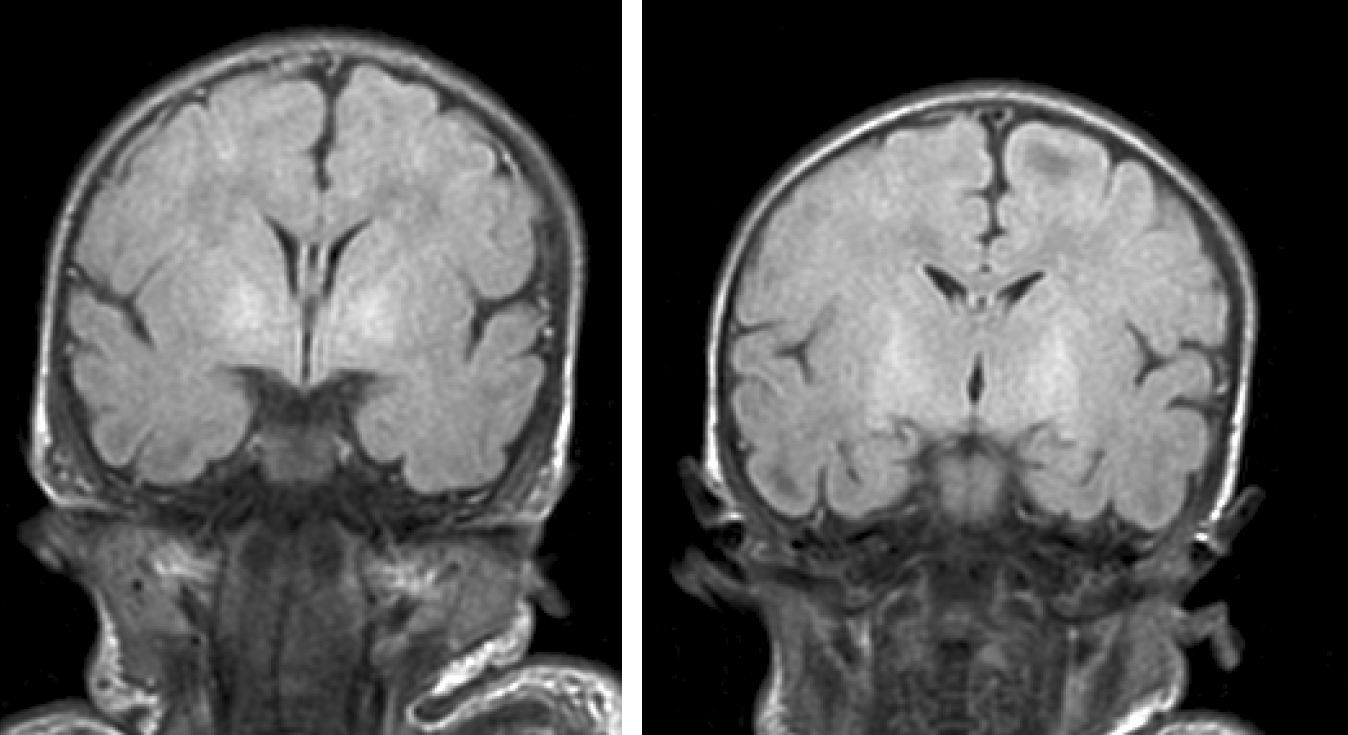
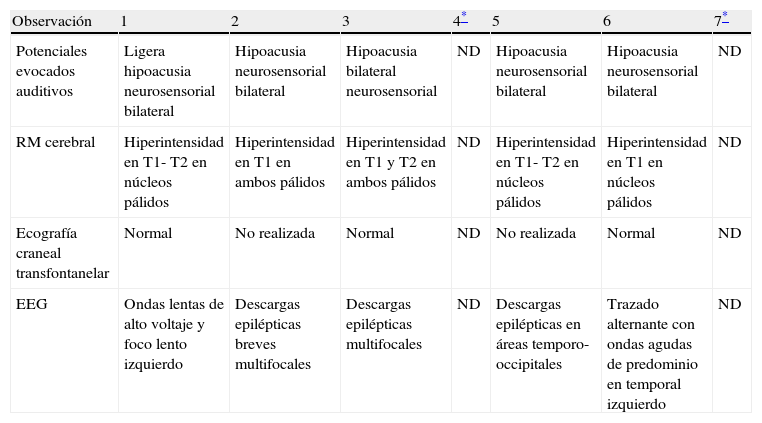
Hallazgos de neuroimagenEn todas las observaciones la ecografía craneal transfontanelar fue normal. La RM craneal se practicó entre los días 6 y 17 de vida. En todos los pacientes se observaron incrementos de señal de forma bilateral en ambos globos pálidos en secuencias T1. En 3 pacientes se observaron igualmente incrementos de señal en núcleo pálido también en secuencias T2 (tabla 2)(figs. 1-3).
Estudios complementarios en período neonatal/primeros meses en los pacientes afectos de kernicterus
| Observación | 1 | 2 | 3 | 4* | 5 | 6 | 7* |
| Potenciales evocados auditivos | Ligera hipoacusia neurosensorial bilateral | Hipoacusia neurosensorial bilateral | Hipoacusia bilateral neurosensorial | ND | Hipoacusia neurosensorial bilateral | Hipoacusia neurosensorial bilateral | ND |
| RM cerebral | Hiperintensidad en T1- T2 en núcleos pálidos | Hiperintensidad en T1 en ambos pálidos | Hiperintensidad en T1 y T2 en ambos pálidos | ND | Hiperintensidad en T1- T2 en núcleos pálidos | Hiperintensidad en T1 en núcleos pálidos | ND |
| Ecografía craneal transfontanelar | Normal | No realizada | Normal | ND | No realizada | Normal | ND |
| EEG | Ondas lentas de alto voltaje y foco lento izquierdo | Descargas epilépticas breves multifocales | Descargas epilépticas multifocales | ND | Descargas epilépticas en áreas temporo-occipitales | Trazado alternante con ondas agudas de predominio en temporal izquierdo | ND |
.Clara hiperintensidad en núcleo pálido y muy discreta en núcleo subtalámico (Obs. 5).")
.")
.")
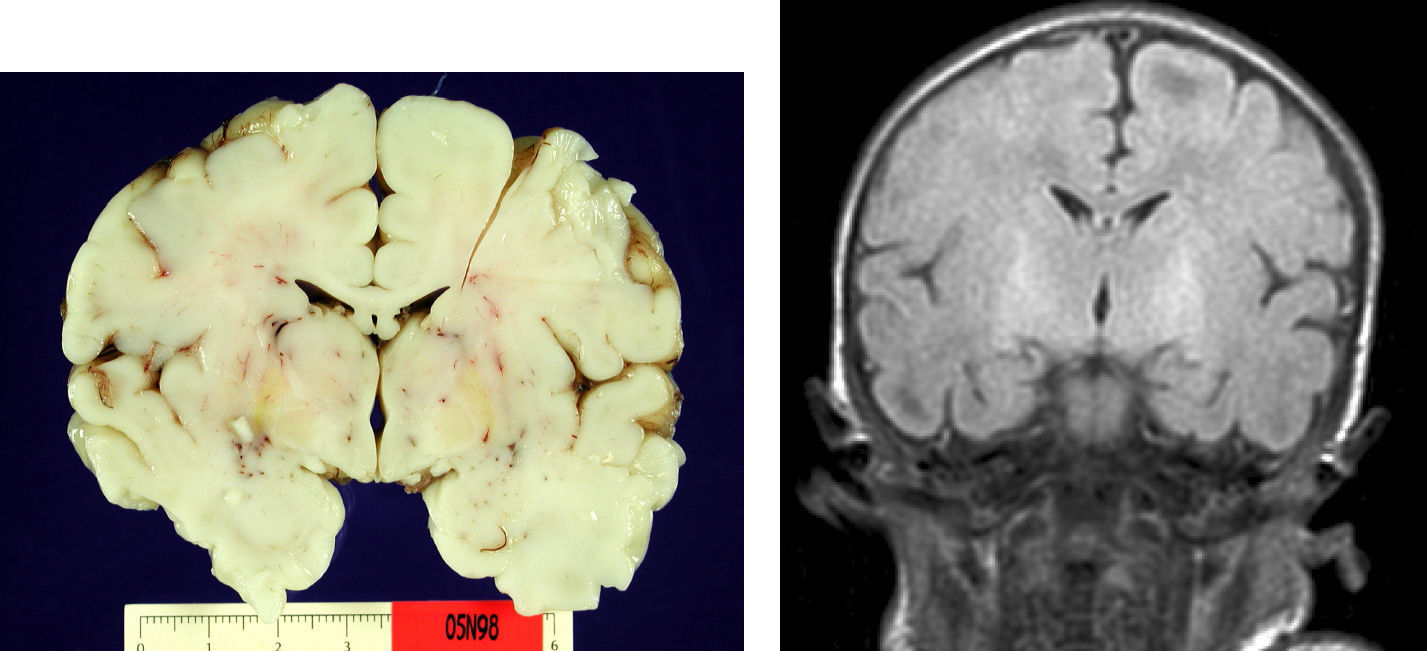
En el paciente que falleció se realizó la necropsia para establecer un diagnóstico etiológico del cuadro. Macroscópicamete, el peso del encéfalo fue de 410 gramos, no presentando alteraciones en las circunvoluciones o en la base del cráneo (sin signos de herniación uncal). Se observó una coloración amarilla en el pedúnculo cerebral izquierdo y en los putámenes, núcleos pálidos y sustancia nigra de los 2 hemisferios cerebrales así como en la zona anterior paramedial de ambos hemisferios cerebelosos (fig. 4).
. RM coronal T2-FLAIR de la misma observación.")
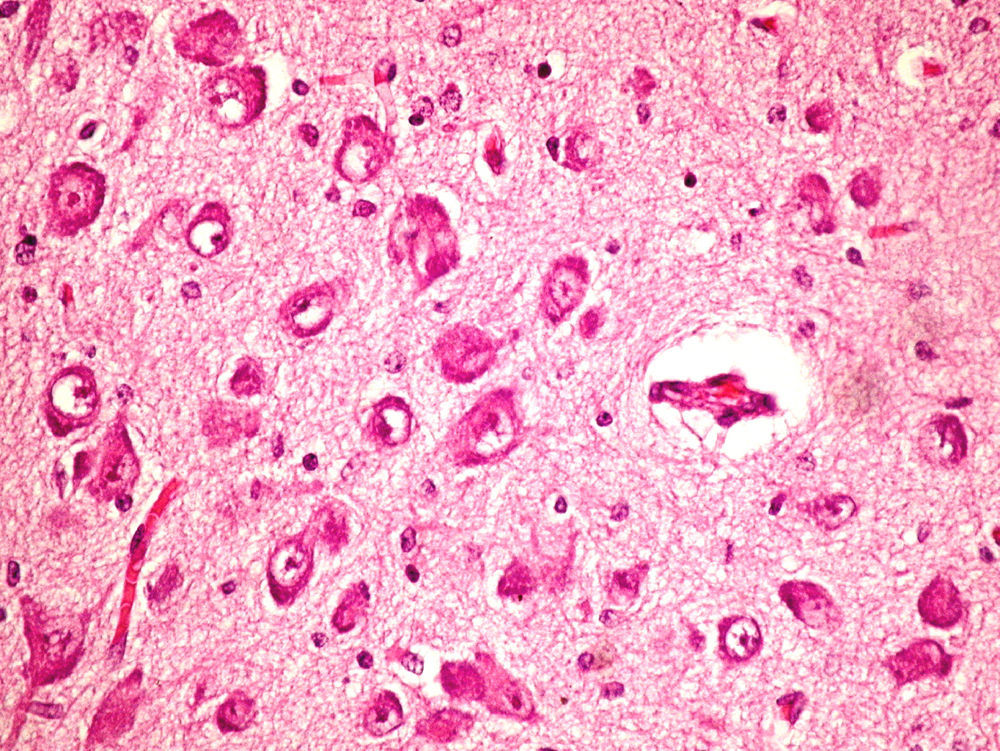
Microscópicamente, a nivel cerebral se observaron cambios compatibles con hipoxia-isquemia (edema, retracción neuronal y picnosis nuclear asociados a astrocitosis) en capas superficiales corticales. En las estructuras profundas (putamen, pálido, núcleo subtalámico, tálamo e hipotálamo así como en los pedúnculos cerebrales) se observaron cambios degenerativos neuronales consistentes en pérdida de la sustancia de Nissl, degeneración eosinófila, vacuolización nuclear y necrosis con pérdida neuronal, todo ello acompañado de astrocitosis y de ocasionales esferoides axonales (fig. 5).
 (Obs. 3). Cambios degenerativos neuronales que muestran pérdida neuronal y astrocitosis.")
A nivel cerebeloso se halló un pigmento ocre en el citoplasma de algunas células de Purkinje, con degeneración y pérdida focal de las mismas en asociación a moderada astrocitosis, estando conservada la capa granular interna.
El estudio inmunohistoquímico en hepatocitos reveló la ausencia total de la enzima bilirrubin-UDP-glucoronosiltransferasa, compatible con el síndrome de Crigler-Najjar tipo I. Sin embargo no se pudo identificar ninguna mutación en el gen UGT1A1.
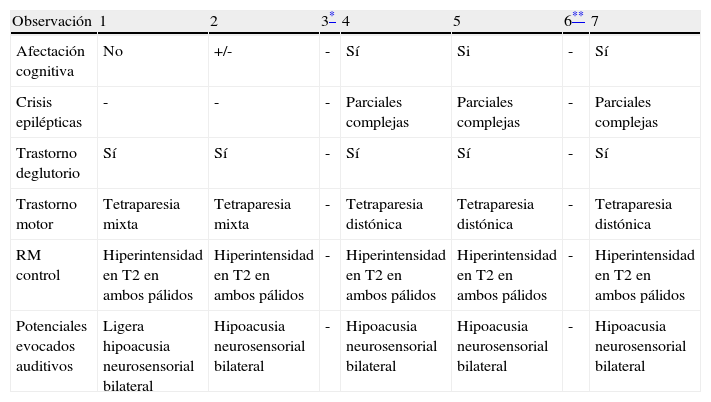
EvoluciónDe los seis supervivientes, todos manifiestaron diverso grado de compromiso neurológico con afectación motora 6/6 tipo tetraparesia mixta en 2 y distónica en 3, hipoacusia neurosensorial 4/5, epilepsia 3/5 y dificultades deglutorias 5/5. La afectación cognitiva estaba presente en 3/5 y solo un paciente asiste a escuela ordinaria con soporte. En todos ellos el cuadro se manifiesta como una encefalopatía estática y la epilepsia tiende a controlarse con FAE (tabla 3).
Evolución y pruebas complementarias realizadas en el seguimiento de los pacientes afectos de kernicterus
| Observación | 1 | 2 | 3* | 4 | 5 | 6** | 7 |
| Afectación cognitiva | No | +/- | - | Sí | Si | - | Sí |
| Crisis epilépticas | - | - | - | Parciales complejas | Parciales complejas | - | Parciales complejas |
| Trastorno deglutorio | Sí | Sí | - | Sí | Sí | - | Sí |
| Trastorno motor | Tetraparesia mixta | Tetraparesia mixta | - | Tetraparesia distónica | Tetraparesia distónica | - | Tetraparesia distónica |
| RM control | Hiperintensidad en T2 en ambos pálidos | Hiperintensidad en T2 en ambos pálidos | - | Hiperintensidad en T2 en ambos pálidos | Hiperintensidad en T2 en ambos pálidos | - | Hiperintensidad en T2 en ambos pálidos |
| Potenciales evocados auditivos | Ligera hipoacusia neurosensorial bilateral | Hipoacusia neurosensorial bilateral | - | Hipoacusia neurosensorial bilateral | Hipoacusia neurosensorial bilateral | - | Hipoacusia neurosensorial bilateral |
Aunque el término kernicterus, que designa la tinción amarilla de los ganglios de la base demostrable solamente en estudios necrópsicos, ha prevalecido por muchos años se considera hoy impreciso y reemplazable por el más adecuado de encefalopatía hiperbilirrubinémica2,3,9. Esta acepción es útil pues indica que diferentes áreas del sistema nervioso central pueden afectarse, y que la “impregnación” o tinción amarilla no es un requisito para que exista disfunción neuronal clínicamente reconocible. En la actualidad pueden ya demostrarse las alteraciones macroscópicas en los ganglios basales en una encefalopatía bilirrubínica mediante RM craneal1,2,6,7.
Se acepta que los niveles séricos de bilirrubina superiores a 20mg/dl aumentan el riesgo de daño neurológico en neonatos de término, pero también se reconoce que el prematuro puede sufrir secuelas importantes con cifras mucho menores, sobre todo si existen factores extra añadidos tales como hipoxia, acidosis, sepsis, hemólisis, poliglobulia o disrupción de la BHE4,5. Cuando se produce una disrupción de la BHE la bilirrubina ligada a la albúmina penetra rapidamente al espacio extracelular cerebral y a su vez la bilirrubina libre produce una grave neurotoxicidad global. En el interior de la neurona, la bilirrubina produce una disrupción del gradiente de protones, interfiere con la homeostasis del calcio, produce hiperexcitabilidad neuronal a traves de los aminoácidos excitadores y un desequilibrio de los neurotrasmisores. Además produce efectos negativos sobre la membrana neuronal, y finalmente interfiere directamente en los procesos oxidativos intramitocondriales, en la producción de energía, conduciendo a la apoptosis y necrosis neuronal2–4. Otros factores a tener en cuenta en la génesis del daño neuronal son el período de exposición a la bilirrubina que varía con la edad gestacional, o el tiempo de exposición a valores altos de bilirrubina4,5,9.
Se ha sugerido recientemente que la BHE, cuando está integra, a través de un sistema de transporte dependiente de ATP actua como una bomba de expulsión de bilirrubina libre y mantiene un gradiente de concentración de bilirrubina libre entre el plasma y LCR10. Existen otros factores que contribuyen al aumento de la bilirrubina como la presencia de sustancias que la desplazan de la unión con la albúmina(las sulfonamidas), o que compiten con los lugares de unión con la albúmina. La existencia de sepsis, hemólisis, acidosis metabólica, y muy especialmente la prematuridad son factores negativos para desarrollar toxicidad por bilirrubina3,10.
El término encefalopatía por hiperbilirrubinemia define un cuadro neurológico que consiste en el depósito de bilirrubina no conjugada en determinadas células cerebrales. Como efecto de este fenómeno, se produce una pigmentación amarillenta y una destrucción de las neuronas afectas. Existen varias zonas que se ven afectadas preferentemente por este fenómeno; éstas son los núcleos pálidos, los núcleos subtalámicos y el hipocampo. Otros lugares comprometidos en menor medida son el tálamo, la sustancia negra, los núcleos cerebelosos y determinados núcleos de los pares craneales. El córtex, la sustancia blanca y el tronco del encéfalo en general no suelen verse afectados4,7,8,11.
Clínicamente la toxicidad por hiperbilirrubinemia puede ser reversible y no dar manifestaciones o que estas sean muy sutiles y aparezcan tardíamente como defectos atencionales, auditivos, o incluso una mínima torpeza motriz2,5 o bien manifestar la clínica neurológica más o menos florida. Las manifestaciones de la encefalopatía por bilirrubina se dividen pues en agudas, crónicas y las ya comentadas manifestaciones sutiles2.
En el período neonatal el cuadro clínico clásico de la encefalopatía hiperbilirrubinémica se manifiesta por signos poco específicos como dificultades de alimentación, irritabilidad, disminución del sensorio, convulsiones o alteraciones del tono muscular (hipertonía o hipotonía, retrocollis, opistótonos), llanto agudo cerebral, ojos en sol poniente, fiebre y coma que puede evolucionar al exitus. En nuestra serie de 7 pacientes cabe señalar que todos ellos desarrollaron clínica neurológica en período neonatal asociada a otras manifestaciones sistémicas propias de su precario estado de salud. En este período los PEATC evidencian alargamiento del intervalo de las ondas I-III y I-V, disminución amplitud de las ondas III y V o incluso abolición de las respuestas. En los 5 pacientes estudiados en perido neonatal o primeros meses de vida mientras permanecían en neonatología se constataron anomalías en los PEATC compatibles. Se ha reportado una mejoria de los PEATC tras la exanguinotransfusión5,12–14. Algunos autores proponen establecer un puntaje pronóstico para la encefalopatía por bilirrubina en fase aguda2,12.
En esta fase aguda la neuroimagen pueden demostrar anomalías en los ganglios basales en la ecografia craneal transfontanelar (que no pudimos apreciar en nuestras observaciones) o muy especialmente alteraciones de señal en la RM craneal en T1 en núcleo pálido y en ocasiones subtalámico6. En todos los pacientes de nuestra serie en los que disponíamos de RM craneal en período neonatal se constató un incremento de señal en T1 en núcleos pálidos y en algún caso en subtalámicos (fig. 1). En 3 pacientes la hiperintensidad también se reflejaba en T2 (figs. 2 y 3).
La forma crónica se caracteriza por una triada clásica con afectacion motora, auditiva y de los movimientos oculares. Estas manifestaciones se han correlacionado perfectamente con la localización topográfica de la impregnación por bilirrubina en ganglios basales (núcleo pálido y subtalámico, cerebelo y tronco cerebral) para las manifestaciones motoras; la impregnación del núcleo del VIII par y del nervio auditivo con la afectación sensorial; y finalmente el compromiso de los núcleos de los pares oculomotores del tronco cerebral para justificar los movimientos oculares anormales propios de la encefalopatía por billirubina2,11,12. Asociado a ello pueden manifestar afectación cognitiva, epilepsia, microcefalia y alteraciones en el esmalte dental2.
La secuela más importante es la afectación motora con una tetraparesia distónica o mixta, que suele mantenerse como una encefalopatía estática y en general pobre respuesta a las medidas de fisioterapia, estimulación o fármacos2,12,15(tabla 3). Solamente un paciente de nuestra serie adquirió la deambulación autónoma a los 4 años.
El defecto neurosensorial es una manifestación habitual y bien conocida de la toxicidad por bilirrubina4,10,12–14. Sin embargo en 1996 Starr et al describen una neuropatía auditiva o también denominada disincronía auditiva, caracterizada por la ausencia de respuesta a los PEATC, con otoemisiones acústicas conservadas y variable grado de compromiso auditivo13. Estos pacientes manifiestan dificultades para la comprensión del lenguaje en ausencia de una importante pérdida auditiva, retardo del lenguaje, problemas de conducta y dificultades en los aprendizajes. Se calcula una prevalencia de este defecto del 5,3 al 14,8% de los neonatos atendidos en cuidados intensivos y especialmente con antecedentes de prematuridad e hiperbilirrubinemia13. La respuesta a los audífonos es escasa, sin embargo los resultados con los implantes cocleares son más esperanzadores16. Las manifestacions oculares (limitación de la mirada al cenit y en ocasiones compromiso de los movimientos oculares horizontales que recuerdan la apraxia oculomotora), no siempre son fáciles de evidenciar en el período neonatal y con el tiempo tienden a mejorar2. En nuestra serie se reportaron, en el período neonatal, anomalías en los movimientos oculares en cuatro pacientes.
La impregnación por bilirrubina suele respetar el córtex cerebral y la sustancia blanca subcortical, por lo cual la afectación cognitiva no es constante; sin embargo en nuestra serie más de la mitad de los pacientes la manifestaba. En otras series el retardo mental y la epilepsia pueden llegar a ser muy importantes2,8.
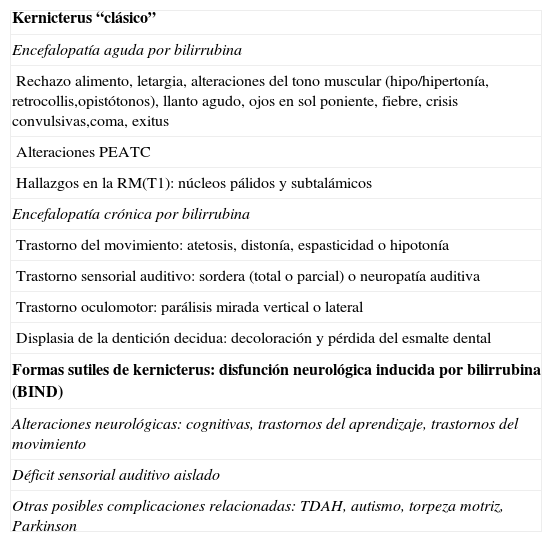
Actualmente se tiende a abandonar el término clásico de kernicterus, considerándose esta entidad como un espectro clínico que puede evolucionar en el tiempo, dentro del cual se englobarían tanto el kernicterus “clásico” como otras formas “sutiles” de kernicterus, tambien conocido como disfunción neurológica inducida por bilirrubina (Bilirrubin Induced Neurological Dysfunction o BIND) (tabla 4)2,13,14. Estas manifestaciones incluyen diversos grados de compromiso neurológico con afectación cognitiva variable, trastornos de los aprendizajes, defectos atencionales movimientos anormales o simplemente la disfunción auditiva ya comentada2,5,11,12.
Espectro clínico de la encefalopatía por hiperbilirrubinemia
| Kernicterus “clásico” |
| Encefalopatía aguda por bilirrubina |
| Rechazo alimento, letargia, alteraciones del tono muscular (hipo/hipertonía, retrocollis,opistótonos), llanto agudo, ojos en sol poniente, fiebre, crisis convulsivas,coma, exitus |
| Alteraciones PEATC |
| Hallazgos en la RM(T1): núcleos pálidos y subtalámicos |
| Encefalopatía crónica por bilirrubina |
| Trastorno del movimiento: atetosis, distonía, espasticidad o hipotonía |
| Trastorno sensorial auditivo: sordera (total o parcial) o neuropatía auditiva |
| Trastorno oculomotor: parálisis mirada vertical o lateral |
| Displasia de la dentición decidua: decoloración y pérdida del esmalte dental |
| Formas sutiles de kernicterus: disfunción neurológica inducida por bilirrubina (BIND) |
| Alteraciones neurológicas: cognitivas, trastornos del aprendizaje, trastornos del movimiento |
| Déficit sensorial auditivo aislado |
| Otras posibles complicaciones relacionadas: TDAH, autismo, torpeza motriz, Parkinson |
El diagnóstico de kernicterus o de encefalopatía por bilirrubina ha sido clínico durante años. Con la mejora de las técnicas de diagnóstico, tanto de laboratorio como de neuroimagen, de los exámenes neurofisiológicos y con la optimización de los tratamientos como la fototerapia o la exanguinotransfusión, se ha conseguido reducir drásticamente el tiempo de diagnóstico pudiendo iniciarse el tratamiento de forma precoz, lo que ha llevado a una disminución clara en la incidencia de esta patología en los últimos años, si bien más recientemente se está observando un cierto repunte, debido entre otros factores a la mayor supervivencia de los recién nacidos pretérmino de muy bajo peso al nacimiento (grandes prematuros)2,5.
En la actualidad la encefalopatía por bilirrubina en el neonato a término o casi a término viene definida no solo por las cifras séricas de bilirrubina, sino por la asociación de una bilirrubina no conjugada > 20mg/dl y la existencia de anomalías neurológicas, ya sean a nivel motor, sensorial, por la presencia de hallazgos sugestivos en la neuroimagen o en PEATC2,5.
Las lesiones patológicas típicas se concretan no solamente por la coloración amarillenta de las estructuras cerebrales que se produce por la exudación regional de bilirrubina (que puede ocurrir sin pérdida neuronal), sino por la asociación además de una afectación neuronal selectiva con gliosis residual de las regiones subtalámicas y de los núcleos pálidos8,9,11. En una de nuestras observaciones, que falleció, se constataron mediante anatomía patológica estos hallazgos. Por otra parte, en nuestra observación los hallazgos compatibles con hipoxia cortical se explican perfectamente por la evolución clínica del paciente con sepsis, coma y exitus secundario a fallo multiorgánico. El paciente permaneció con ventilación mecánica durante todo el ingreso. Es interesante el hecho de que hasta la fecha existen pocos trabajos que correlacionen los hallazgos macroscópicos con las imágenes de RM8,9,11,12.
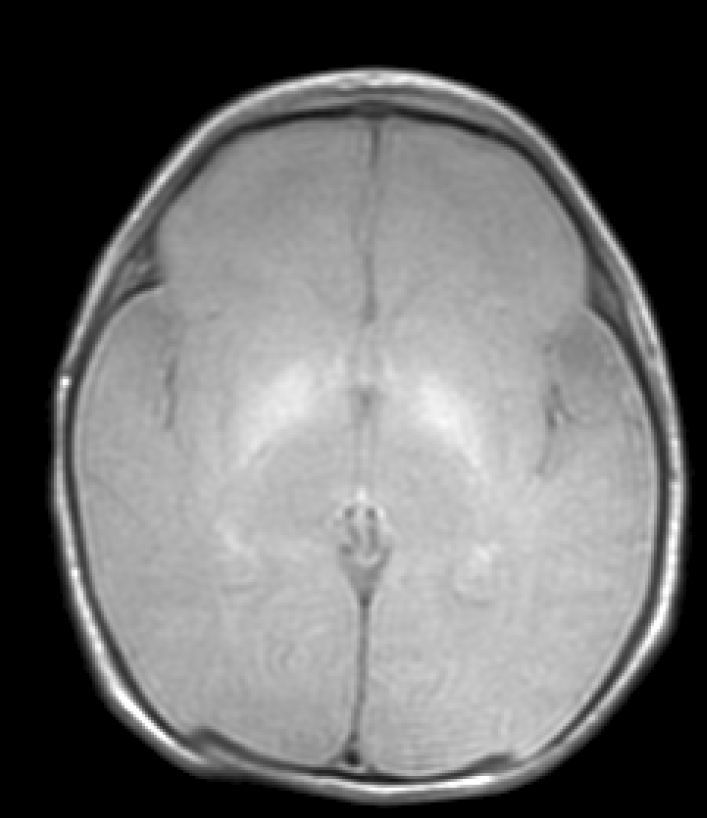
Con la aparición de la RM, las lesiones cerebrales propias de este síndrome se han identificado y descrito en neonatos a término2,12,15,17–20. Existe una secuencia cronológica de las lesiones del kernicterus detectables por RM craneal. En una primera fase (fase aguda) se puede detectar un incremento de señal en secuencias T1 en los núcleos pálidos y en ocasiones en núcleos subtalámicos. Estas alteraciones han sido ya descritas en neonatos pretérmino o a término hiperbilirrubinémicos con 5 días de vida15. Se piensa que estos hallazgos reflejarían la reacción astroglial inmediata frente al insulto, edema o incluso por la propia presencia de la bilirrubina6,7,19,20. En una segunda fase (transicional), se produce una atenuación de la hiperseñal en T1 a lo largo de las 2-3 semanas de vida hasta normalizarse. En algunos pacientes se observa de forma simultánea la aparición de una hiperseñal en T2 en la misma localización, incluso puede extenderse a cerebelo y tronco2. Se han descrito aumentos de señal en T2 desde los días 6 y 8 de vida7,12. Tres de nuestros pacientes presentaban cambios de señal en secuencias T2 (a los 6 y 17 días de vida), mientras que los otros 3 no manifestaban estos cambios a los 7 días de vida. Todos ellos presentaron una evolución desfavorable con exitus o secuelas importantes (tabla 3). La evolución de la neuroimagen en todas nuestras observaciones es bastante similar, con práctica desaparición de la hiperseñal en T1 y aparición de la hiperseñal en T2 en núcleos pálidos y en dos observaciones en núcleos subtalámicos (figs. 2,4).
Finalmente, en una tercera etapa (fase crónica) se produce un aumento de señal en T2 que persiste a lo largo de la vida (fig. 3), habiéndose encontrado en niños de hasta 12 años19. Esta señal traduciría los cambios de densa gliosis fibrilar pobre en contenido celular que aparece en la fase final, perfectamente descrita desde los años 708,9.
Los hallazgos de neuroimagen en todos los pacientes no siempre pasan por las tres fases. Se ha publicado una serie de 6 pacientes con anomalías en las RM iniciales consistentes en hiperseñal exclusivamente en secuencias T1 en núcleos pálidos con normalización de las imágenes en RM posteriores de control18,19. Cuatro de estos niños (4/6) tuvieron un desarrollo normal15. Hay que insistir aquí de nuevo en que no todos los pacientes con hiperbilirrubinemia, manifestaciones neurológicas en período neonatal y neuroimagen positiva van a desarrollar la clinica de la enfermedad5. Este hecho tiene gran importancia para emitir un pronóstico en período neonatal o incluso para proceder a medidas terapeuticas más o menos agresivas. No obstante este hecho es excepcional y cuando la clínica neonatal, los parámetros bioquímicos, de neuroimagen y de PEATC son muy evidentes las probabilidades de desarrollar la enfermedad son muy elevadas.
Por todo ello creemos que la RM craneal es un instrumento útil en el período neonatal para demostrar la toxicidad de la bilirrubina en sistema nervioso central. La asociación en el período neonatal de hiperbilirrubinemia con sintomatología clínica acompañante y cambios de señal en la RM (hiperseñal en ambos globos pálidos inicialmente en T1 y en T2 en etapas posteriores) junto a la alteraciones en los PEATC nos permiten el diagnóstico, en general orientar el pronóstico e iniciar al tratamiento rehabilitador.
Hasta el momento, la única forma de prevenir estos desórdenes la constituye una determinación de las cifras de bilirrubinemia sérica ante la mínima sospecha y el tratamiento oportuno si procede. La aplicación de protocolos internacionales consensuados de tratamiento de la hiperbilirrubinemia con fototerapia y exanguinotransfusión y otras medidas opcionales como la seroalbumina al 5% como preexanguinotransfusión, los inhibidores de hemoxigenasa, los inductores enzimáticos de la conjugación, la inducción de la excreción intestinal de bilirrubina, los inhibidores de la betaglucuronidasa y la quelación intestinal entre otros, deben contribuir a reducir/ minimizar la toxicidad de la bilirrubina a nivel del sistema nervioso central en estas edades tempranas21–23.
Como conclusión señalar que a pesar de los avances científicos seguimos viendo pacientes con secuelas de hiperbilirrubinemia neonatal y que los mecanismos fisiopatológicos se conocen mejor pero no en su totalidad. En los últimos diez años hemos observado un repunte de la incidencia de casos de encefalopatía por hiperbilirrubinemia que podemos atribuir en parte al incremento de las complicaciones propias del gran prematuro/neonato de alto riesgo cuando hace unos años estos pacientes no sobrevivían al período neonatal. En segundo lugar el aumento de la población inmigrante procedente de países menos desarrollados que no disponen de medios para monitorizar la bilirrubina y para tratar las cifras elevadas contribuye a este incremento de la casuística. Gracias a las nuevas técnicas de neuroimagen pueden diagnosticarse con precisión las lesiones en ganglios basales, cerebelo y tronco atribuibles a la hiperbilirrubinemia y responsables de la encefalopatía. Finalmente señalar que no se puede descartar que muchos pacientes afectos de hipoacusia neurosensorial y en los que se desconoce la causa puedan ser atribuibles a la hiperbilirrubinemia del período neonatal o incluso frente algunas manifestaciones neurológicas menores en las que no encontramos una explicación etiológica razonable.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Trabajo presentado a la reunion anual de la AINP en Tuxla-Gutierrez, Mexico.

