




Consideramos encefalopatías prenatales las que tienen datos clínicos o prenatales de encefalopatía antes del nacimiento. Afectan a un número importante de niños controlados en las consultas de neuropediatría. Pueden ser disruptivas (por problemas vasculares durante el embarazo, drogas, tóxicos o infecciones congénitas), y genéticamente determinadas. Incluimos casos de trastorno del espectro autista y retardo mental sin historia de sufrimiento perinatal o postnatal.
Material y métodosSe revisa nuestra experiencia en el diagnóstico etiológico de las encefalopatías prenatales durante los últimos 19 años. Se analizan los estudios realizados en los casos sin diagnóstico etiológico.
ResultadosEn el periodo de estudio de 19 años y 5 meses, en la base de datos de neuropediatría figuran 11.910 niños. Tienen establecido el diagnóstico de encefalopatía prenatal 1596 (13,5%). De ellos no tienen diagnóstico etiológico preciso 1.307 niños (81,4%) pese a habérseles realizado múltiples estudios complementarios, fundamentalmente bioquímicos, genéticos y de neuroimagen.
DiscusiónMuchos de los niños incluidos en este estudio presentan enfermedades raras, estén o no identificadas, que demandan crecientemente un diagnóstico precoz. Enfermedades peroxisomales, lisosomales, mitocondriales, defectos congénitos de la glucosilación, entre otras enfermedades metabólicas hereditarias, infecciones congénitas, cromosomopatías y genopatías, pueden ser indistinguibles clínicamente y necesitan estudios específicos para su identificación. Un diagnóstico precoz precisa estrategias de estudios sistemáticos de forma escalonada, priorizando las enfermedades que tienen posibilidades terapéuticas y en muchos casos es necesaria también una aproximación individualizada. Creemos que las ventajas potenciales del diagnóstico precoz, incluido el ahorro de más pruebas, y la prevención, probablemente sobrepasan el gasto financiero.
We examine those prenatal encephalopathies with clinical or neuroimaging data of encephalopathy before the birth. They affect a significant number of children seen by paediatric neurologists. They can be of disruptive origin (due to vascular problems, drugs, toxins or congenital infections), and genetically determined. We include cases of autism spectrum disorder and mental retardation with no history of perinatal of postnatal damages.
Material and methodsWe analysed our 19 year neuro-paediatric data base in search of prenatal encephalopathies and their diagnostic origin. We also analyse the studies made in the cases with a diagnosis of unknown origin.
ResultsThe 19 year period of study in the data base included 11,910 children, and 1596 (13.5%) were considered as prenatal encephalopathies; 1307 children (81.4%) had a diagnosis of unknown origin, despite many investigations being done in a large number of them.
DiscussionMost of the children included in this study suffer a rare disease, and whether they are identified or not, they increasingly require an early diagnosis. Peroxisomal, mitochondrial, lysosomal diseases, carbohydrate glycosylation deficiency syndrome and other inborn error of metabolism, congenital infections and genetic encephalopathies, can be clinically indistinguishable in early life and require specific studies to identify them. Early diagnosis requires strategies using step-wise systematic studies, giving priority to those diseases that could be treated, and in many cases using an individualised approach. We believe that the potential benefits of early diagnosis, including savings on further studies, genetic counselling and prenatal diagnosis, overcome the financial costs.
El proceso asistencial más frecuente del neuropediatra empieza con un motivo de consulta, a partir del cual trata de establecer un diagnóstico que puede tener hasta cuatro diferentes niveles:
- 1.
Ubicación topográfica del problema: encefalopatía (que, a su vez, puede ser con afectación exclusiva o asociada de hemisferios cerebrales, cerebelo, línea media, etc.); debe plantearse siempre la posible afectación vías visuales y auditivas; mielopatía; trastorno de la unidad neuromuscular (afectación de asta anterior, nervio periférico [axonal o mielínica]), unión neuromuscular o músculo.
- 2.
Localización temporal del origen del problema: problemas prenatales (genéticamente determinados o disruptivos), perinatales o posnatales (accidente, infección…). Algunos problemas no tienen una localización temporal concreta: algunos casos de determinadas enfermedades metabólicas hereditarias (EMH) y síndromes neurocutáneos asocian alteraciones del sistema nervioso central que ya empiezan intraútero.
- 3.
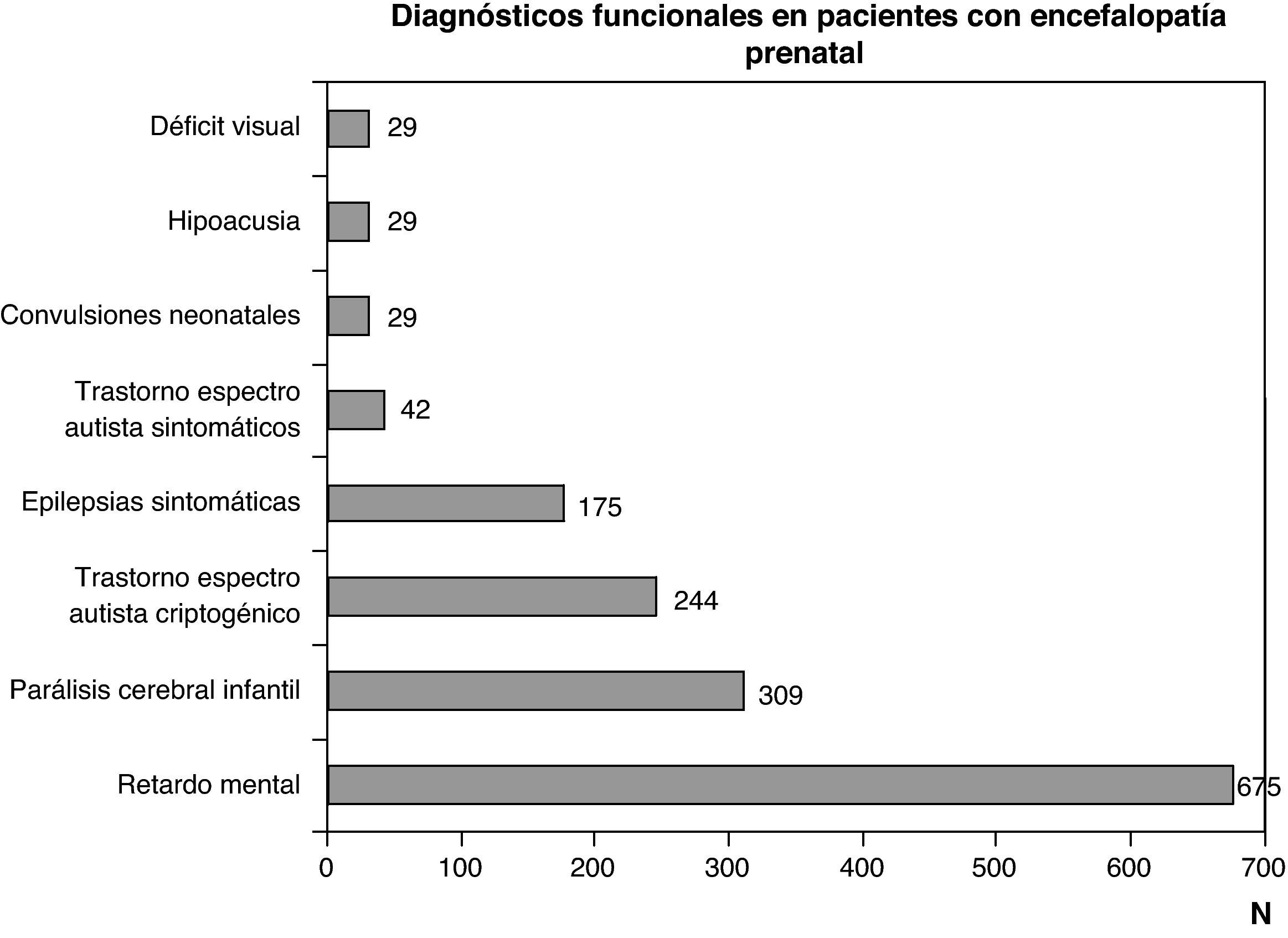
Diagnóstico funcional: problemas motores por disfunciones a cualquier nivel del sistema nervioso central, problemas cognitivos y comportamentales, y epilepsia por disfunciones encefálicas y problemas sensoriales (visión o audición) que obedecen a encefalopatías o a trastornos de los órganos de la vista o de la audición. Los diagnósticos funcionales más frecuentes son parálisis cerebral infantil, deficiencia mental, espectro autista y epilepsia. Todos ellos son consecuencia de encefalopatías, pero pueden obedecer a diferentes etiologías, tanto prenatales como perinatales y posnatales. Un problema motor puro puede deberse tanto a una encefalopatía como a un trastorno de la médula espinal o de la unidad neuromuscular. Una encefalopatía puede manifestarse por alteraciones aisladas o asociadas motoras, cognitivas, comportamentales o sensoriales (déficit visual o hipoacusia). El déficit visual y la hipoacusia pueden deberse exclusivamente a alteraciones de los órganos de los sentidos, que a su vez pueden asociarse a encefalopatía o a alteraciones a otros niveles del sistema nervioso.
- 4.
Diagnóstico etiológico. Es el que permite cerrar el proceso diagnóstico, valorar las posibilidades terapéuticas, establecer un pronóstico y un riesgo de repetición en la familia. Los avances en el ámbito terapéutico y de prevención y diagnóstico prenatal se acompañan de una creciente demanda de diagnóstico precoz.
Con frecuencia no es posible identificar todos estos niveles de diagnóstico. Además, los problemas muestran grados diversos de severidad, pueden estar aislados o asociados varios de ellos, y son evolutivos, y las repercusiones pueden ser permanentes o transitorias.
Un número importante de los niños controlados en las consultas de neuropediatría presentan encefalopatías prenatales, entendidas como producidas antes del nacimiento del niño. Pueden ser disruptivas (por problemas vasculares durante el embarazo, por drogas o tóxicos o por infecciones congénitas) y genéticamente determinadas, incluidas las EMH.
Desde esta perspectiva, se revisa la experiencia en el diagnóstico etiológico de las encefalopatías prenatales de nuestra unidad de neuropediatría durante los últimos 19 años.
Material y métodosSe analizan los casos catalogados de encefalopatía prenatal, independientemente de su gravedad, procedentes de la base de datos de neuropediatría desde su inicio el 15 de mayo de 1990 hasta el 6 de octubre de 20091–5.
El término encefalopatía se ha utilizado obedeciendo a su significado etimológico de padecimiento encefálico, independientemente de su carácter difuso o localizado y de las repercusiones clínicas. El diagnóstico de encefalopatía prenatal se ha establecido considerando criterios clínicos y/o de neuroimagen. Apoyan el origen prenatal de una encefalopatía datos como la existencia de polihidramnios, rasgos dismórficos faciales y malformaciones extraneurológicas asociadas, así como la no evidencia de noxa perinatal o posnatal. Se consideran también encefalopatías prenatales los casos de deficiencia mental y de autismo criptogénicos. Es diagnóstico de encefalopatía prenatal la identificación por neuroimagen de agenesia de cuerpo calloso, de trastornos de la migración neuronal o de otras anomalías prenatales. Quedan excluidos los casos sin evidencia de encefalopatía prenatal de cuadros regresivos, síndromes neurocutáneos y enfermedades de la unidad neuromuscular.
Utilizamos el término criptogénico, de acuerdo con su significado etimológico, aplicándolo a los problemas de causa no conocida.
Se recogen los casos con diagnóstico etiológico totalmente establecido y los que entendemos no precisan estudios genéticos o bioquímicos, como en los casos de encefalopatía asociada al mielomeningocele (hidrocefalia, anomalía de Chiari II y disgenesia de cuerpo calloso entre otras) o síndromes neurocutáneos con evidencia de encefalopatía prenatal.
Por otro lado, se analizan los casos de encefalopatía prenatal de causa no aclarada, sus diagnósticos funcionales, de localización topográfica y tipo de alteración estructural en su caso, así como algunos de los estudios realizados en ellos en el proceso diagnóstico. No se han podido recoger algunas pruebas realizadas por no haberlas introducido en la base de datos hasta recientemente: serie esquelética, ecografía abdominal, estudio cardiológico, oligosacáridos y glucosaminoglucanos en orina.
ResultadosEn el periodo de estudio de 19 años y 5 meses, en la base de datos de neuropediatría figuran 11.910 niños. Tienen establecido el diagnóstico de encefalopatía prenatal 1.596 (13,5%). De ellos no tienen diagnóstico etiológico preciso 1.307 niños (81,4%). Del total de la base de datos hemos considerado que tienen enfermedad rara 1.480 niños, sabedores de que muchos de los que no tienen diagnóstico establecido también tienen enfermedades raras.
En la tabla 1 se recogen los diagnósticos etiológicos establecidos en los 289 niños en los que ello ha sido posible.
Diagnósticos establecidos en las encefalopatías prenatales.
| Cromosomopatías (n=107) |
| Síndrome de Down (n=33) |
| Síndrome X- frágil (n=15) |
| Síndrome de Angelman (n=8) |
| Enfermedad de Prader Willi (n=7) |
| Deleción subtelomérica (n=7) |
| Síndrome de Edwards (n=7) |
| Síndrome de Patau (n=1) |
| Síndrome de Williams Beuren (n=1) |
| Otras cromosomopatías (n=28) |
| Malformaciones del sistema nervioso central (n=59) |
| Mielomeningocele con afectación cerebral (n=31) |
| Microcefalia genética (n=15) |
| Agiria/lisencefalia con mutación gen LIS1 (n=3) |
| Síndrome de Joubert (n=2) |
| Síndrome de Walker Warburg (n=1) |
| Enfermedad metabólica hereditaria (n=34) |
| Enfermedad mitocondrial* (n=16) |
| Déficit congénito de la glucosilación (n=3) |
| Hiperglicinemia no cetósica (n=3) |
| Síndrome de Cockayne (n=2) |
| Gangliosidosis GM1 (n=1) |
| Enfermedad de Hurler (n=1) |
| Defecto beta-oxidación mitocondrial (n=1) |
| Distrofia muscular congénita deficiente en merosina (n=1) |
| Enfermedad de Pelizaeus Mezbacher (n=1) |
| Enfermedad peroxisomal (variante Zellweger) (n=1) |
| Smith Lemli Opitz (n=1) |
| Enfermedad de jarabe de arce (n=1) |
| Aciduria 2-metil 3-hidroxibutiril CoA deshidrogenasa (n=1) |
| Déficit de biotinidasa (n=1) |
| Encefalopatía disruptiva (n=63) |
| Infecciones congénitas (n=32) |
| Encefalopatía teratogénica (n=21): 16 SAF |
| Vascular: 9 gemelar (n=10) |
| Síndrome dismórfico con retardo mental (n=20) |
| Síndrome neurocutáneo (n=10) |
| Distrofia miotónica congénita (n=2) |
| Distrofia muscular de Duchenne con retardo mental (n=1) |
SAF: síndrome alcohólico fetal.
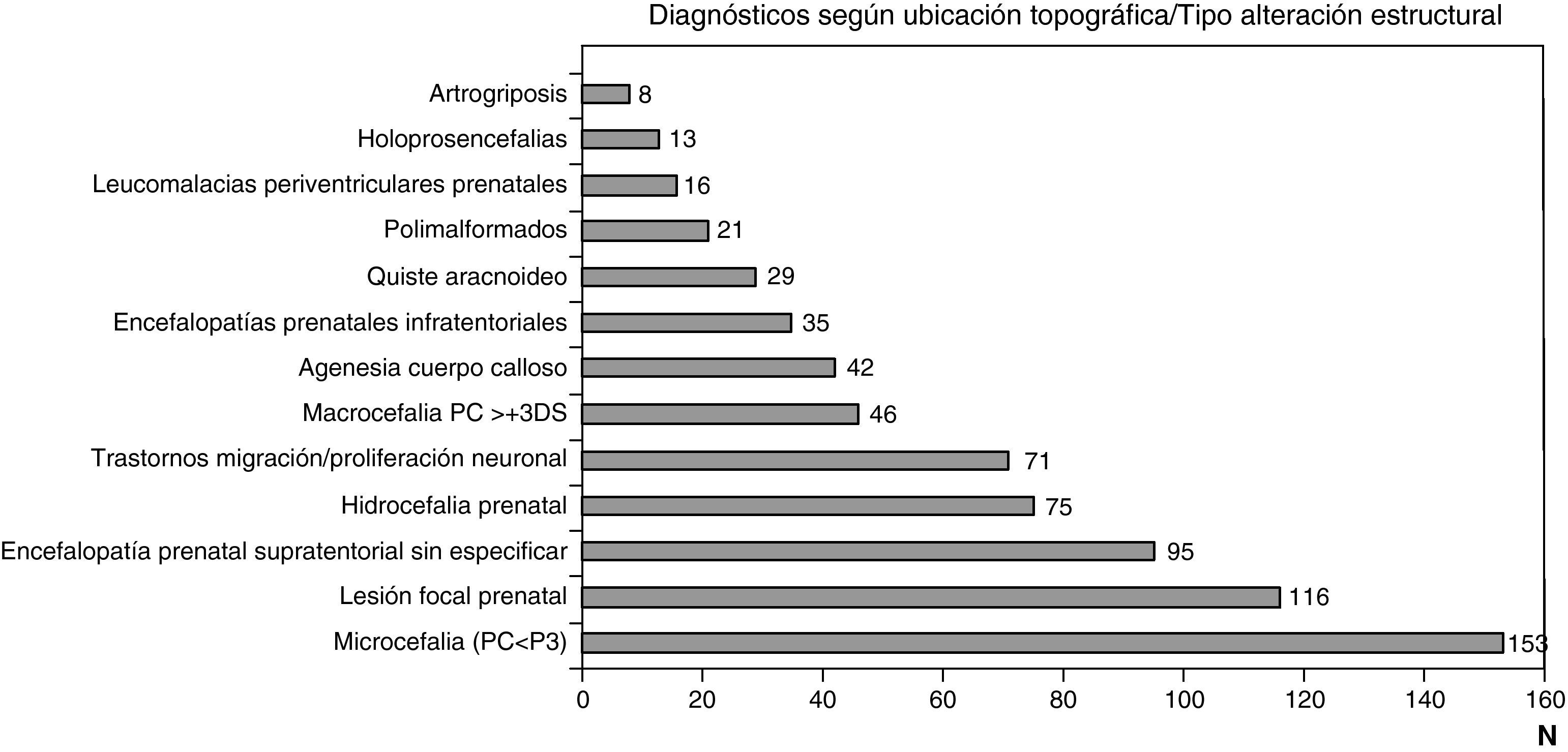
Las figuras 1 y 2 recogen los diagnósticos más importantes funcionales, de localización topográfica y de tipo de alteración estructural asignados a los 1.307 niños con encefalopatía prenatal sin diagnóstico etiológico establecido.


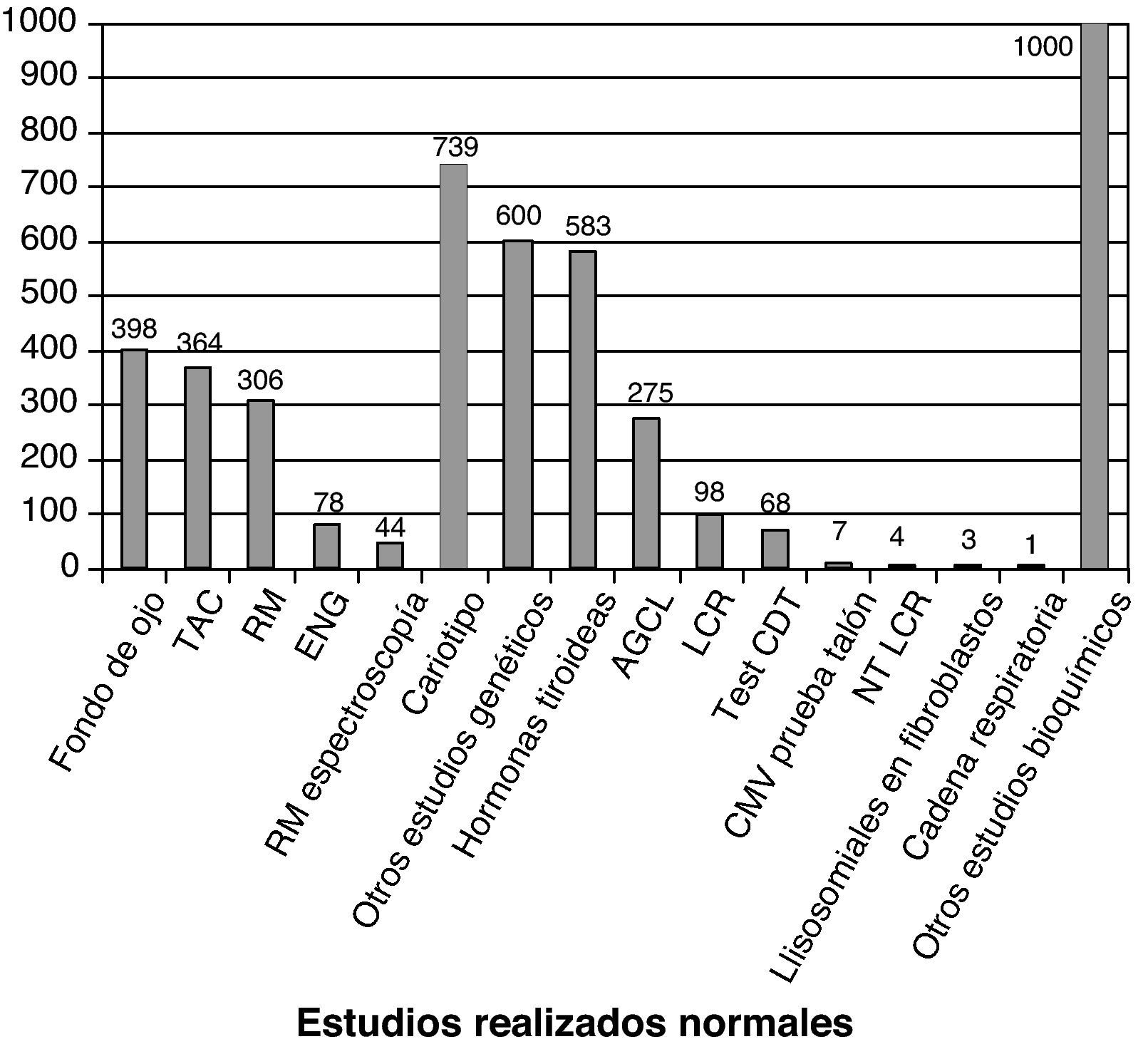
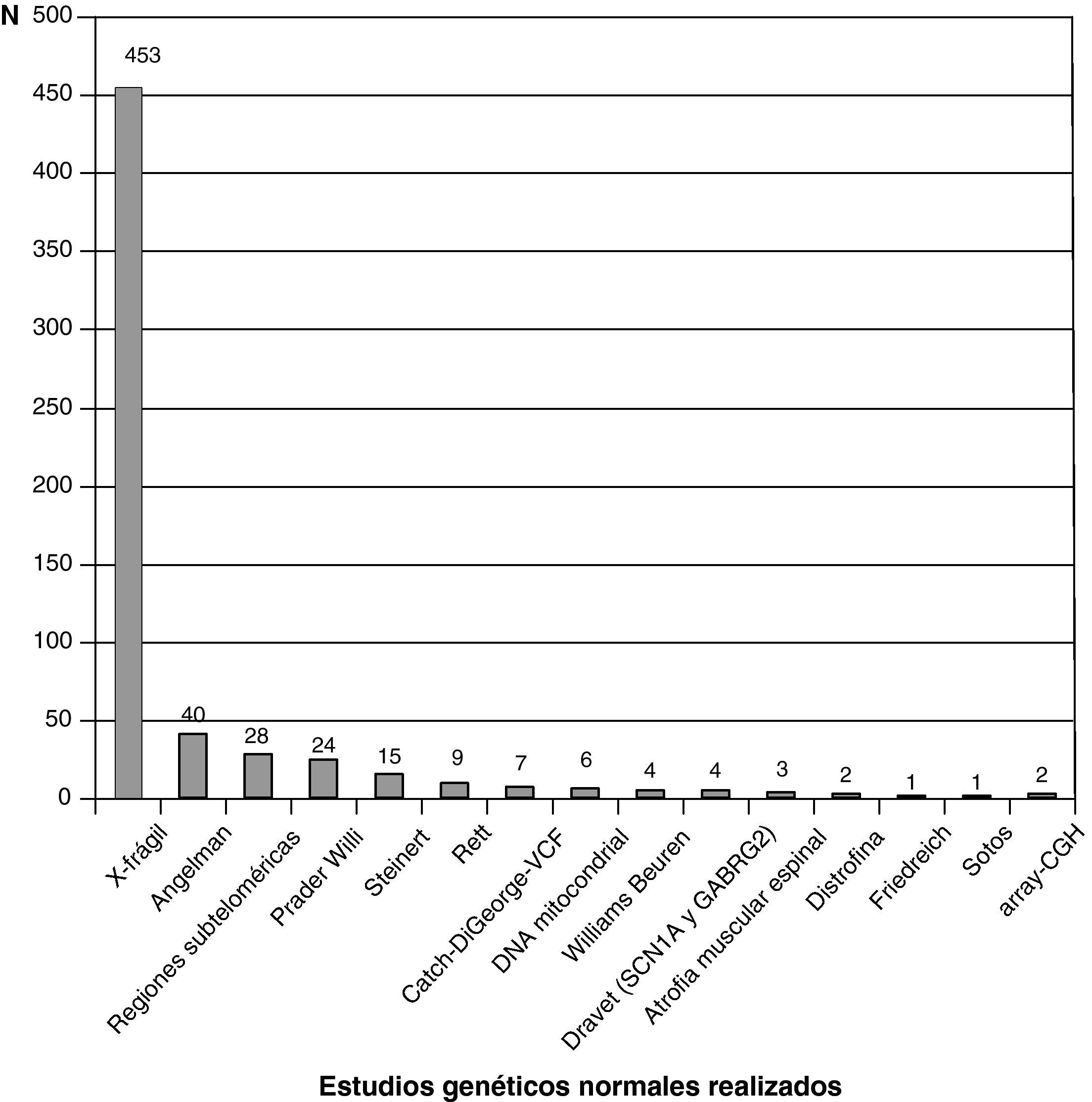
La figura 3 recoge los estudios complementarios practicados y que han resultado normales en los 1.307 niños sin diagnóstico etiológico establecido. La figura 4 recoge los estudios genéticos, diferentes de cariotipo, normales realizados. Se obtuvieron resultados anormales que no permitieron establecer el diagnóstico etiológico al menos 1 vez en 320 niños de TC craneal, 251 niños de RM cerebral, 35 niños de fondo de ojo y 6 niños de electroneurograma.
. Si está alterado se determina patrón sérico de sialotransferrinas.")
Estudios normales realizados a los 1307 niños sin diagnóstico etiológico establecido. Se recoge número de niños con al menos un estudio normal realizado. AGCL: ácidos grasos de cadena larga; NTLCR: neurotransmisores en el líquido cefalorraquídeo; test de CDT: porcentaje de transferrina deficientemente carboxilada; es útil en la identificación de los defectos congénitos de la n-glucosilación (CDG). Si está alterado se determina patrón sérico de sialotransferrinas.

Admitimos que el concepto definido de encefalopatía prenatal es amplio y en él pueden tener cabida otros trastornos que implican la participación de áreas disfuncionales cerebrales, como los trastornos de la lectoescritura o el trastorno por déficit de atención e hiperactividad; en ellos, la afectación es menor y se acepta, en general, la poca rentabilidad de los estudios complementarios. En este trabajo hemos pretendido analizar nuestros datos reales sobre estudios realizados y diagnósticos etiológicos establecidos en un grupo amplio y poco homogéneo de problemas. Los estudios se han realizado, por tanto, de forma no homogénea en todos los pacientes, siguiendo los criterios clínicos y disponibilidades que han evolucionado a lo largo del periodo analizado, y siguen en permanente adaptación.
Las exigencias sociosanitarias aumentan con el acceso generalizado a la información, la participación activa de los pacientes y sus familiares en el control de la enfermedad, el aumento de las posibilidades diagnósticas y de diagnóstico prenatal y con las cada vez mejores opciones presentes o futuras de tratamiento. Las posibilidades terapéuticas de algunas enfermedades lisosomales, mitocondriales y peroxisomales (la adrenoleucodistrofia ligada a X, por ejemplo), incrementan enormemente la trascendencia de un diagnóstico precoz.
Recientemente se ha aprobado la «Estrategia de enfermedades raras del Sistema Nacional de Salud»6, en total sintonía con la Recomendación del Consejo de Europa relativa a una acción en el ámbito de las enfermedades raras7. La segunda línea estratégica es la prevención y la detección precoz.
Muchos de los niños incluidos en este estudio (tabla 1 y figs. 1 y 2) están afectados de enfermedades raras, estén o no identificadas. En el momento actual, las enfermedades raras se benefician de un gran ritmo en los avances en su conocimiento y todas ellas poseen unas características generales comunes. Son muy poco frecuentes, incluso agrupadas todas ellas; en su mayoría son hereditarias, lo que implica necesidad de asesoramiento genético y estudio de familiares potencialmente afectados, así como estrategias de diagnóstico prenatal; tienen una gran complejidad diagnóstica; muchas de ellas son muy graves, crónicas y discapacitantes, y existe una gran complejidad de las opciones terapéuticas. Como consecuencia de todo ello se genera una gran exigencia a los profesionales involucrados y la necesidad de equipos multidisciplinarios en su manejo.
Destacamos el elevado porcentaje de niños con encefalopatía prenatal que no tienen un diagnóstico etiológico (81%), pese al elevado número de estudios realizados (figs. 3 y 4). Sin embargo, dichos estudios son necesarios para identificar muchos de los casos con diagnóstico etiológico establecido. Es parte de nuestro trabajo controlar niños sin diagnóstico y no es fácil encontrar un equilibrio entre hacer muchos estudios para obtener pocos diagnósticos y conformarse con la ausencia de diagnóstico.
En la tabla 1 se refleja que en 10 de 16 diagnósticos de enfermedad mitocondrial dicho diagnóstico no es de certeza, por tratarse de déficits parciales de la cadena respiratoria, en casos de clínica inespecífica y sin otros apoyos diagnósticos. Esa falta de certeza nos llevó a realizar menos estudios tras el auge diagnóstico de enfermedades mitocondriales que vivimos hace unos años. En 2009 nos propusimos hacer un mayor esfuerzo por diagnosticar enfermedades genéticas y EMH.
Las encefalopatías prenatales, genéticamente determinadas y disruptivas, incluidas muchas EMH de inicio prenatal, pueden ser indistinguibles clínicamente. Enfermedades peroxisomales, lisosomales, mitocondriales y defectos congénitos de la glucosilación entre otras EMH, infecciones congénitas, cromosomopatías y genopatías precisan estudios específicos para su identificación8,10.
Dada la inespecificidad de signos y síntomas de muchas encefalopatías prenatales y EMH de inicio prenatal o precoz, un diagnóstico precoz precisa estrategias de estudios sistemáticos de forma escalonada, que deben estar en permanente actualización9.
La neuroimagen es útil en el estudio de encefalopatías prenatales; la RM cerebral es la que ofrece mayor rentabilidad11,12. Pese a la necesidad de anestesia, se ha impuesto a la TC craneal además por la ausencia de radiaciones ionizantes13, circunstancia tanto más importante cuanta menor es la edad del paciente.
En la sistemática de estudio de las encefalopatías prenatales y EMH de inicio prenatal o precoz se incluye la valoración de posibles alteraciones asociadas a nivel oftalmológico, cardiológico, abdominal, nefrourológico, esquelético, muscular y del sistema nervioso periférico. El electroneurograma es especialmente útil en los primeros años de la vida, donde puede faltar evidencia clínica de neuropatía y en procesos que asocian afectación de primera motoneurona como algunas leucodistrofias en los que la afectación clínica piramidal puede prevalecer sobre la neuropatía9.
Las posibilidades diagnósticas aumentan con la disponibilidad de estudios, especialmente bioquímicos y genéticos14,15. Además de los recogidos en las figuras 3 y 4, deben hacerse en algunos casos estudios de infección congénita en periodo neonatal y determinaciones de oligosacáridos y glucosamicoglucanos, y de creatina y guanidino acetato en orina.
En esta revisión probablemente faltan estudios genéticos realizados en pacientes sin diagnóstico, presumiblemente genético en muchos casos, lo que puede obedecer en parte a la poca rentabilidad del cariotipo convencional. Los estudios genéticos están en avance permanente y a un ritmo de difícil adaptación. Apenas hemos empezado a realizar estudios de deleciones subteloméricas cuando surgen los estudios array-CGH. La disponibilidad y la rentabilidad del cariotipo de alta resolución y del array-CGH, están cambiando la orientación diagnóstica. Tal vez en un futuro cercano empezaremos por los estudios genéticos y sólo ante su normalidad se plantearán otros exámenes complementarios
Algunas EMH tienen marcadores biológicos que orientan el diagnóstico. El patrón sérico de sialotransferrinas o la prueba de CDT (porcentaje de transferrina deficientemente carboxilada) identifican la mayoría de los defectos congénitos de la glucosilación. El patrón de ácidos grasos de cadena larga se encuentra en la mayoría de las enfermedades peroxisomales. Otros datos bioquímicos permiten identificar muchas EMH, como enfermedad de Menkes, síndrome de Smith-Lemli-Opitz y síndrome de Lesh-Nyhan, entre otras. Sin embargo, hay casos en los que tras la realización de estudios complementarios seriados e incluso repetidos no es posible determinar si se trata de una EMH, y puede estar justificado realizar el estudio ordenado, según la mayor sospecha clínica, de la cadena respiratoria mitocondrial en músculo y piel y de determinadas actividades enzimáticas lisosomales y defectos de piruvato deshidrogenasa en cultivo de fibroblastos.
Las estrategias deben atenerse al principio de justicia y deben aplicarse en todos los casos en los que estén indicadas16,17. Dichas estrategias no excluyen en muchos casos el planteamiento individualizado de cada paciente.
El diagnóstico en algunos casos asocia posibilidades terapéuticas, casi siempre permite establecer el riesgo de repetición, con frecuencia asocia posibilidades de diagnóstico prenatal y preimplantacional, y siempre permite dar respuestas a los familiares y también a nosotros los médicos, incluidos el pronóstico y los planteamientos en algunos casos de medidas de prolongación de la vida, más difíciles de establecer en ausencia de un diagnóstico etiológico establecido.
Es difícil establecer límites y no disponemos de «evidencias» que justifiquen dichos estudios. Creemos que las ventajas potenciales del diagnóstico precoz, incluido el ahorro de más pruebas, y la prevención probablemente sobrepasan el gasto financiero9. Obviamente, en las estrategias diagnósticas deben priorizarse las enfermedades que tienen posibilidades terapéuticas8,9,14.
En cualquier caso, es necesario el esfuerzo de adaptación a las crecientes exigencias de diagnóstico precoz. Lamentablemente, las posibilidades de adaptación de los neuropediatras y expertos en EMH como colectivo son cada vez más limitadas: somos pocos y con elevada carga de trabajo asistencial y de adaptación a las crecientes demandas. La oferta pública de empleo, que prima la antigüedad, está en total confrontación con el mejor servicio público, que debe buscar los mejores profesionales con el perfil idóneo. Dichos profesionales precisan de una formación específica, remunerada, y de forma ideal con dedicación completa. Tenemos pocas posibilidades de crecimiento y de mejora por la ausencia de reconocimiento de las especialidades pediátricas y de planteamiento y planificación de las autoridades sanitarias.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Presentado en el VII Congreso Nacional de Errores Congénitos del Metabolismo. Bilbao, 22-23 de octubre de 2009.

