




El retraso global del desarrollo (RGD) y la discapacidad intelectual (DI) son un motivo de consulta frecuente en la consulta de Neuropediatría. Actualmente, la hibridación genómica comparada constituye una de las principales técnicas aplicadas al diagnóstico de esta patología. Resulta útil determinar qué características fenotípicas se asocian a obtener un resultado etiológico en el test genético.
MétodosSe llevó a cabo un estudio ciego pormenorizado de las características clínicas, antropométricas y morfológicas de 80 individuos afectos de DI no explicada y se analizó cuales estaban asociadas a obtener un resultado etiológico en el array-CGH.
ResultadosEl resultado del array fue patológico en un 27,5% de los casos. Los factores que se asociaron estadísticamente a tener una prueba de array-CGH patológica fueron los antecedentes familiares de DI/RGD (OR: 12,1), la presencia de malformaciones congénitas (OR: 5,33), más de 3 rasgos dismórficos faciales (OR: 20,9) y la hipotonía periférica (OR: 3,25).
ConclusionesNuestros hallazgos coinciden con otras series publicadas. Por lo tanto, asumimos que la probabilidad de encontrar variación en el número de copias de significado patológico mediante array-CGH aumenta si alguna de las características anteriores está presentes en individuos afectos de DI/RGD.
Global developmental delay (GDD) and intellectual disability (ID) are frequent reasons for consultation in paediatric neurology departments. Nowadays, array comparative genomic hybridisation (array-CGH) is one of the most widely used techniques for diagnosing these disorders. Our purpose was to determine the phenotypic features associated with pathological results in this genetic test.
MethodsWe conducted a blind study of the epidemiological, clinical, anthropometric, and morphological features of 80 patients with unexplained ID to determine which features were associated with pathological results in array-CGH.
ResultsPathological results were found in 27.5% of the patients. Factors associated with pathological results in array-CGH were a family history of GDD/ID (OR = 12.1), congenital malformations (OR = 5.33), having more than 3 facial dysmorphic features (OR = 20.9), and hypotonia (OR = 3.25).
ConclusionsOur findings are consistent with those reported by other published series. We therefore conclude that the probability of having pathological results in array-CGH increases with the presence of any of the features mentioned above in patients with ID/GDD.
La discapacidad intelectual (DI) se caracteriza por limitaciones significativas tanto en el funcionamiento intelectual como en el adaptativo y se origina con anterioridad a los 18 años. Para los niños menores de 5 años se usa el término retraso global del desarrollo (RGD) en aquellos que no cumplen los hitos esperados para su edad en las áreas de desarrollo social, motor o intelectual (con principal relevancia del lenguaje)1-3. Aproximadamente el 50% de los casos a nivel global no tienen un diagnóstico etiológico establecido4,5. Las causas más frecuentes identificadas dentro del campo de la genética han sido las mutaciones puntuales en genes conocidos y las alteraciones cromosómicas6,7. La técnica array-comparative genomic hybridization (CGH) identifica tanto excesos (duplicaciones) como defectos (deleciones) de material genético y constituye uno de los principales test diagnósticos en niños con trastornos del neurodesarrollo. Cuando se aplica al estudio de la DI el rendimiento diagnóstico varía desde un 10 a un 30%, según los criterios de selección de la muestra utilizados8,9. El objetivo de nuestro estudio es conocer qué características clínicas, antropométricas y morfológicas aparecen con mayor frecuencia en individuos afectados de DI/RGD con un resultado diagnóstico en la prueba del array-CGH.
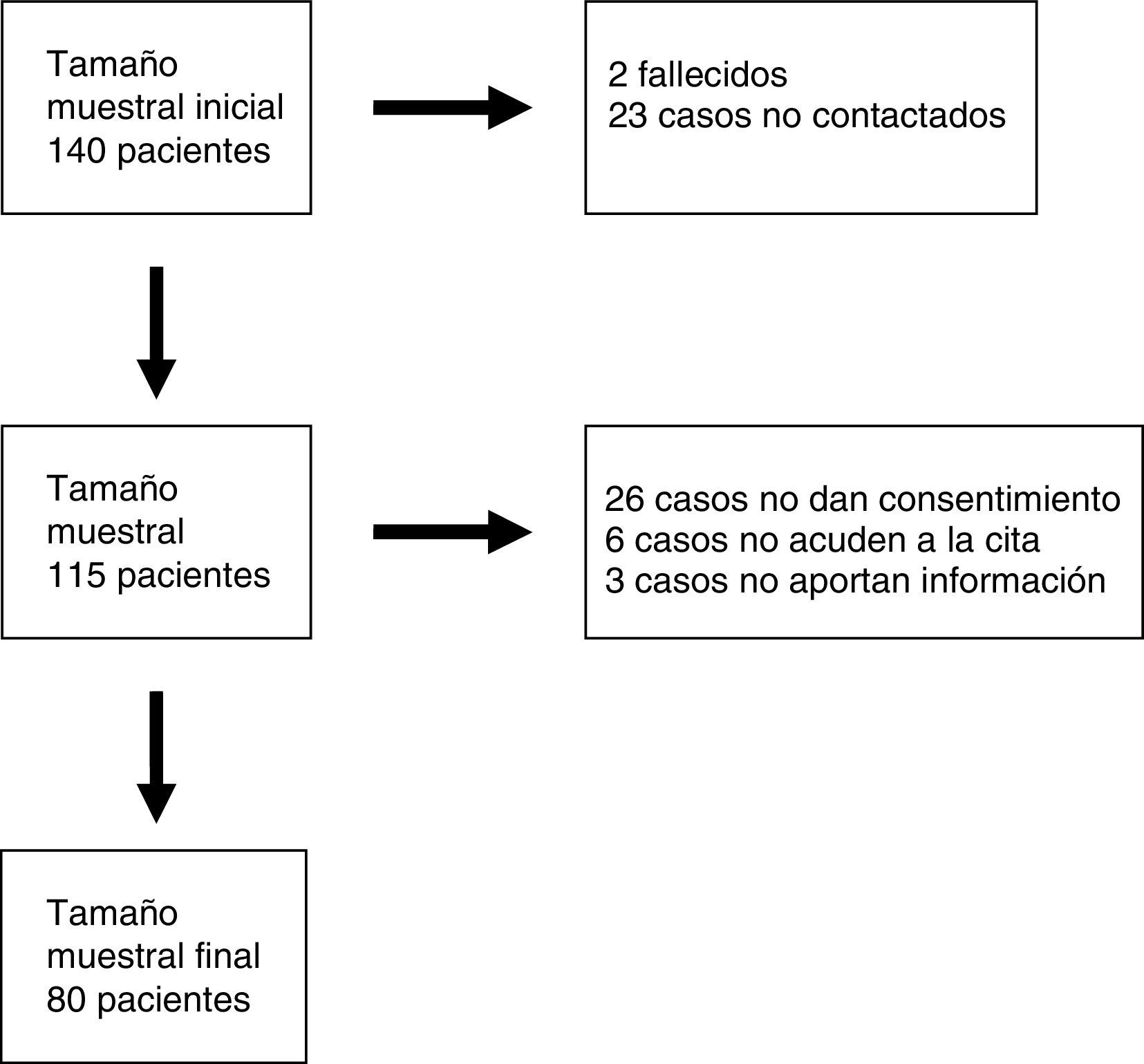
Pacientes y métodosLos criterios de selección de la muestra fueron: a) estar en seguimiento en la consulta de Neuropediatría del Hospital Infantil Miguel Servet de Zaragoza; b) edad comprendida entre los 12 meses y los 18 años; c) estar afectados de DI o RGD sin diagnóstico etiológico establecido, y d) haberse realizado la técnica array-CGH y haber recibido el resultado antes del 1 de diciembre del 2011. El número total de individuos que cumplían estos criterios fue de 140. El periodo de investigación fue de 2 años y se desarrolló por un único investigador.
Se diagnosticó de RGD a aquellos pacientes que con una edad inferior a 5 años presentaban una alteración en el neurodesarrollo en 2 o más áreas según la escala de desarrollo Denver o Llevant. Asimismo, se diagnosticó de DI a aquellos pacientes que a una edad igual o superior a 5 años disponían de un estudio de cociente intelectual<70. En aquellos casos que no se disponía de un estudio neuropsicológico, la DI se definió en función del tipo de escolarización: educación especial o con adaptación curricular significativa.
Método: se envió una carta informativa de las características del estudio a las familias y se concertó una cita presencial por vía telefónica. El día de la cita se firmó el consentimiento informado (repetido) y se registraron las principales variables demográficas, perinatales, clínicas, morfológicas y antropométricas de cada uno de los individuos sin conocer el resultado del test genético mediante entrevista clínica y exploración física pormenorizada. A posteriori, se revisaron la historia clínica y el resultado del array-CGH clasificando a los individuos en 3 grupos: resultado normal, resultado patológico o resultado incierto. Para llevar a cabo esta clasificación se consultaron las bases de datos Database of Genomic Variants (DGV), International Standard Cytogenomic Arrayconsortium Databases (ISCA), Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER) y Online Mendelian Inheritance in Man (OMIM).
El estudio se llevó a cabo siguiendo las normas deontológicas reconocidas por la Declaración de Helsinki (59.ª Asamblea General, Seúl, Corea, octubre del 2008)10. El proyecto fue revisado y aprobado por el Comité Ético de Investigación Clínica de Aragón. Asimismo, se obtuvo el consentimiento informado de cada uno de los pacientes.
El análisis genético se realizó a partir de ADN extraído de linfocitos de sangre periférica, mediante el sistema de extracción automatizado EZ1 (Quiagen GmbH, Hilden, Alemania). Las muestras se remitieron para su análisis mediante hibridación genómica comparada sobre array a 2 laboratorios: Qgenomics y Genycell Biotech. La hibridación se llevó a cabo con un microarray de 60.000 sondas oligonucleotídicas, repartidas por todo el genoma, con mayor cobertura en regiones pericentroméricas, subteloméricas e implicadas en trastornos genómicos recurrentes.
Una vez recogidas las variables de estudio, se comprobaron la normalidad de la distribución con la prueba Shapiro-Wilk y la homogeneidad de varianzas mediante la prueba de Levene. La asociación entre los factores estudiados y el resultado de la prueba array-CGH se investigó mediante pruebas de contraste de hipótesis, con comparación de proporciones cuando ambas variables eran cualitativas (chi al cuadrado para variables que cumplen supuesto de normalidad y prueba exacta de Fisher cuando no la cumplen) y comparaciones de medias cuando una de ellas sea cuantitativa, t de Student y ANOVA para una distribución normal, y el test de la U de Mann-Whitney para distribuciones no normales. Se comparó la distribución de las características de los individuos con un resultado normal del array-CGH con los que presentaban un resultado patológico. Se excluyó del análisis a los individuos con un resultado incierto y el caso n.° 15 que simultáneamente presentaba un síndrome X frágil y cuyas características podían actuar como factor de confusión.
Para el desarrollo del modelo de regresión logística, se procedió a un cribado de las variables explicativas, a partir del análisis de regresiones univariantes, seleccionando así los potenciales factores pronósticos que se debían incluir en el modelo multivariante (valores p < 0,25)11,12. Con las variables resultantes o las que por motivos clínicos debían estar presentes en el modelo final se exploraron los diferentes modelos multivariantes con los métodos de inclusión y exclusión secuencial, fijando los valores p de significación para la inclusión o exclusión de variables en 0,1 y 0,2, respectivamente. En todos los casos se fijó el nivel de significación estadística para valores de p < 0,05. El análisis estadístico fue realizado mediante el programa informático IBM SPSS Statistics 21 (SPSS Inc., Chicago, EE. UU.).
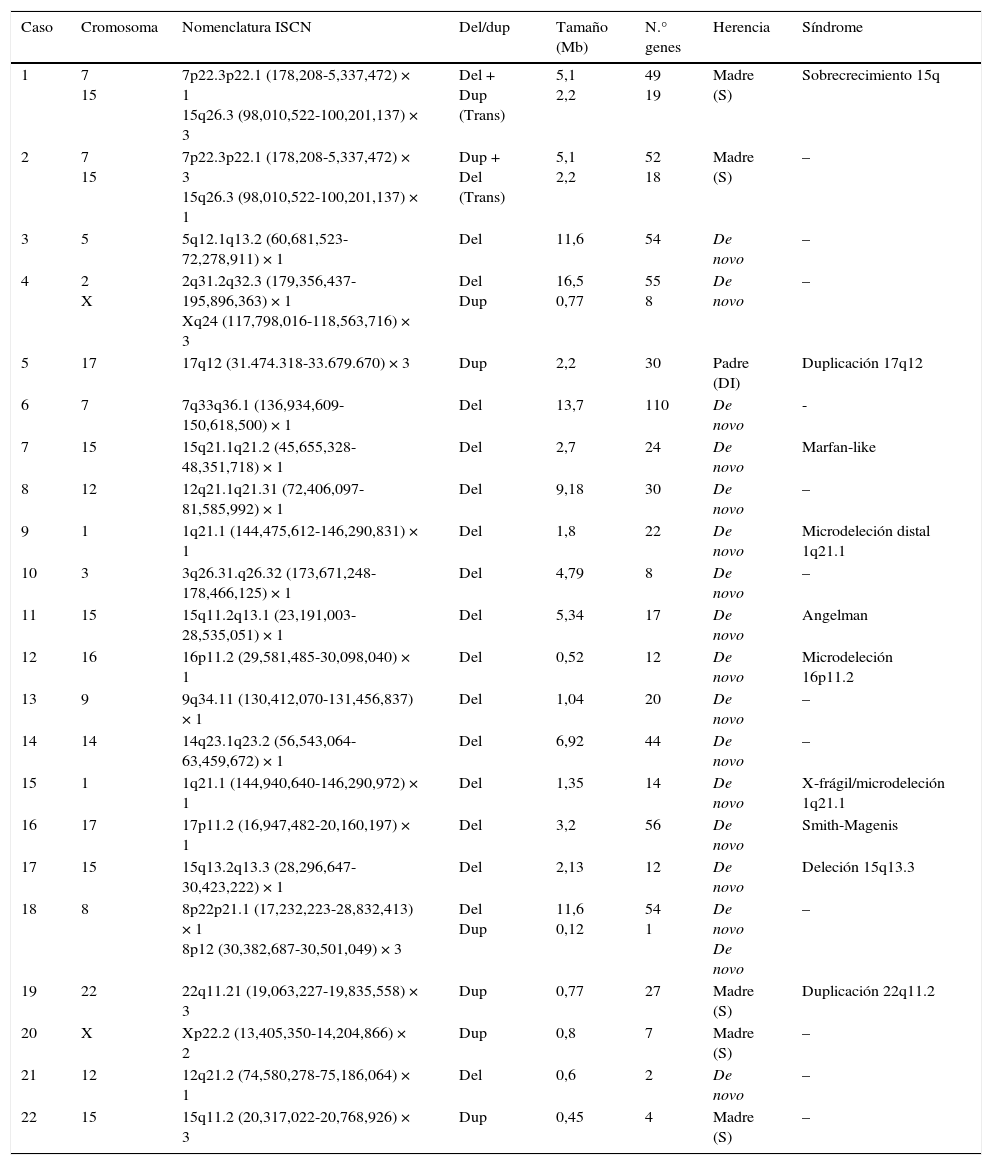
ResultadosOchenta individuos terminaron todas las fases del estudio (fig. 1). Se encontraron variantes en el número de copias (CNV) en un 40% de los casos y se consideraron patológicas un 27,5% (tabla 1). Con respecto al tipo de desequilibrio cromosómico, en un 62,5% de los casos se detectaron deleciones, en un 6,25% translocaciones no equilibradas y en un 31,25% duplicaciones. El tamaño de las deleciones osciló entre 0,06 y 16,5 Mb y el de las duplicaciones entre 0,08 y 2,2 Mb. En un 51,6% de los análisis las alteraciones cromosómicas se produjeron de novo. En los casos heredados de un progenitor, en 7 fue del padre (un individuo afectado, resto sanos) y en 8 de la madre (todas sanas, aunque 2 de ellas portadoras de una translocación equilibrada).

Características genéticas de los casos con resultado del array-CGH patológico
| Caso | Cromosoma | Nomenclatura ISCN | Del/dup | Tamaño (Mb) | N.° genes | Herencia | Síndrome |
|---|---|---|---|---|---|---|---|
| 1 | 7 15 | 7p22.3p22.1 (178,208-5,337,472) × 1 15q26.3 (98,010,522-100,201,137) × 3 | Del + Dup (Trans) | 5,1 2,2 | 49 19 | Madre (S) | Sobrecrecimiento 15q |
| 2 | 7 15 | 7p22.3p22.1 (178,208-5,337,472) × 3 15q26.3 (98,010,522-100,201,137) × 1 | Dup + Del (Trans) | 5,1 2,2 | 52 18 | Madre (S) | – |
| 3 | 5 | 5q12.1q13.2 (60,681,523-72,278,911) × 1 | Del | 11,6 | 54 | De novo | – |
| 4 | 2 X | 2q31.2q32.3 (179,356,437-195,896,363) × 1 Xq24 (117,798,016-118,563,716) × 3 | Del Dup | 16,5 0,77 | 55 8 | De novo | – |
| 5 | 17 | 17q12 (31.474.318-33.679.670) × 3 | Dup | 2,2 | 30 | Padre (DI) | Duplicación 17q12 |
| 6 | 7 | 7q33q36.1 (136,934,609-150,618,500) × 1 | Del | 13,7 | 110 | De novo | - |
| 7 | 15 | 15q21.1q21.2 (45,655,328-48,351,718) × 1 | Del | 2,7 | 24 | De novo | Marfan-like |
| 8 | 12 | 12q21.1q21.31 (72,406,097-81,585,992) × 1 | Del | 9,18 | 30 | De novo | – |
| 9 | 1 | 1q21.1 (144,475,612-146,290,831) × 1 | Del | 1,8 | 22 | De novo | Microdeleción distal 1q21.1 |
| 10 | 3 | 3q26.31.q26.32 (173,671,248-178,466,125) × 1 | Del | 4,79 | 8 | De novo | – |
| 11 | 15 | 15q11.2q13.1 (23,191,003-28,535,051) × 1 | Del | 5,34 | 17 | De novo | Angelman |
| 12 | 16 | 16p11.2 (29,581,485-30,098,040) × 1 | Del | 0,52 | 12 | De novo | Microdeleción 16p11.2 |
| 13 | 9 | 9q34.11 (130,412,070-131,456,837) × 1 | Del | 1,04 | 20 | De novo | – |
| 14 | 14 | 14q23.1q23.2 (56,543,064-63,459,672) × 1 | Del | 6,92 | 44 | De novo | – |
| 15 | 1 | 1q21.1 (144,940,640-146,290,972) × 1 | Del | 1,35 | 14 | De novo | X-frágil/microdeleción 1q21.1 |
| 16 | 17 | 17p11.2 (16,947,482-20,160,197) × 1 | Del | 3,2 | 56 | De novo | Smith-Magenis |
| 17 | 15 | 15q13.2q13.3 (28,296,647-30,423,222) × 1 | Del | 2,13 | 12 | De novo | Deleción 15q13.3 |
| 18 | 8 | 8p22p21.1 (17,232,223-28,832,413) × 1 8p12 (30,382,687-30,501,049) × 3 | Del Dup | 11,6 0,12 | 54 1 | De novo De novo | – |
| 19 | 22 | 22q11.21 (19,063,227-19,835,558) × 3 | Dup | 0,77 | 27 | Madre (S) | Duplicación 22q11.2 |
| 20 | X | Xp22.2 (13,405,350-14,204,866) × 2 | Dup | 0,8 | 7 | Madre (S) | – |
| 21 | 12 | 12q21.2 (74,580,278-75,186,064) × 1 | Del | 0,6 | 2 | De novo | – |
| 22 | 15 | 15q11.2 (20,317,022-20,768,926) × 3 | Dup | 0,45 | 4 | Madre (S) | – |
Del: deleción; DI: discapacidad intelectual; Dup: duplicación; ISCN: International System for Human Cytogenetics; (S): sana; Trans: translocación.
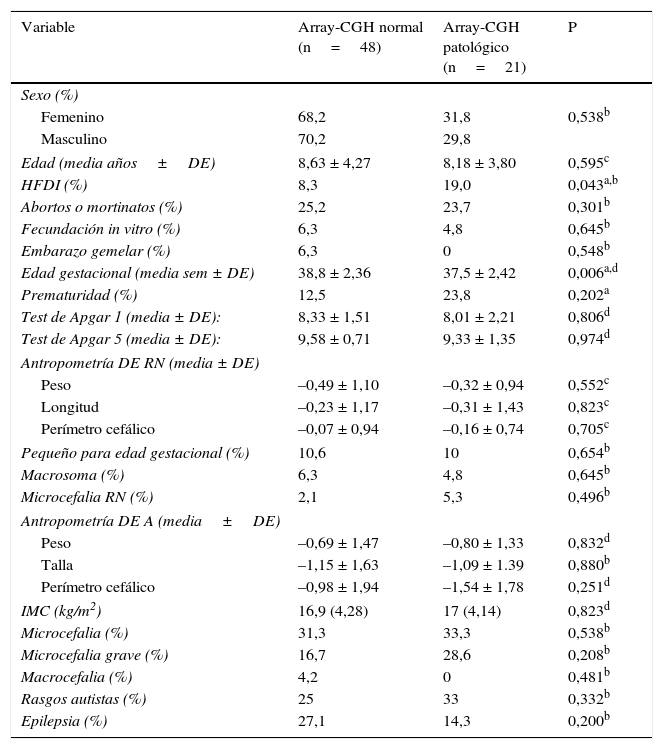
Con respecto a las principales características demográficas, perinatales, antropométricas y clínicas se encontró asociación entre los antecedentes familiares de DI y el resultado patológico del array (p = 0,043) (OR: 12,1; IC del 95%: 3,18-45,9). La media de la edad gestacional fue inferior en el grupo con array-CGH patológico (p = 0,006), aunque no se encontraron diferencias significativas con respecto a la prematuridad en ambos grupos (tabla 2).
Comparación de las características demográficas, perinatales, antropométricas y clínicas
| Variable | Array-CGH normal (n=48) | Array-CGH patológico (n=21) | P |
|---|---|---|---|
| Sexo (%) | |||
| Femenino | 68,2 | 31,8 | 0,538b |
| Masculino | 70,2 | 29,8 | |
| Edad (media años±DE) | 8,63 ± 4,27 | 8,18 ± 3,80 | 0,595c |
| HFDI (%) | 8,3 | 19,0 | 0,043a,b |
| Abortos o mortinatos (%) | 25,2 | 23,7 | 0,301b |
| Fecundación in vitro (%) | 6,3 | 4,8 | 0,645b |
| Embarazo gemelar (%) | 6,3 | 0 | 0,548b |
| Edad gestacional (media sem ± DE) | 38,8 ± 2,36 | 37,5 ± 2,42 | 0,006a,d |
| Prematuridad (%) | 12,5 | 23,8 | 0,202a |
| Test de Apgar 1 (media ± DE): | 8,33 ± 1,51 | 8,01 ± 2,21 | 0,806d |
| Test de Apgar 5 (media ± DE): | 9,58 ± 0,71 | 9,33 ± 1,35 | 0,974d |
| Antropometría DE RN (media ± DE) | |||
| Peso | –0,49 ± 1,10 | –0,32 ± 0,94 | 0,552c |
| Longitud | –0,23 ± 1,17 | –0,31 ± 1,43 | 0,823c |
| Perímetro cefálico | –0,07 ± 0,94 | –0,16 ± 0,74 | 0,705c |
| Pequeño para edad gestacional (%) | 10,6 | 10 | 0,654b |
| Macrosoma (%) | 6,3 | 4,8 | 0,645b |
| Microcefalia RN (%) | 2,1 | 5,3 | 0,496b |
| Antropometría DE A (media±DE) | |||
| Peso | –0,69 ± 1,47 | –0,80 ± 1,33 | 0,832d |
| Talla | –1,15 ± 1,63 | –1,09 ± 1.39 | 0,880b |
| Perímetro cefálico | –0,98 ± 1,94 | –1,54 ± 1,78 | 0,251d |
| IMC (kg/m2) | 16,9 (4,28) | 17 (4,14) | 0,823d |
| Microcefalia (%) | 31,3 | 33,3 | 0,538b |
| Microcefalia grave (%) | 16,7 | 28,6 | 0,208b |
| Macrocefalia (%) | 4,2 | 0 | 0,481b |
| Rasgos autistas (%) | 25 | 33 | 0,332b |
| Epilepsia (%) | 27,1 | 14,3 | 0,200b |
DE A: antropometría actual expresada en desviaciones estándar referidas a su sexo y edad; DE RN: antropometría en el recién nacido expresada en desviaciones estándar referidas a su sexo y edad gestacional; IMC: índice de masa corporal (kg/m2).
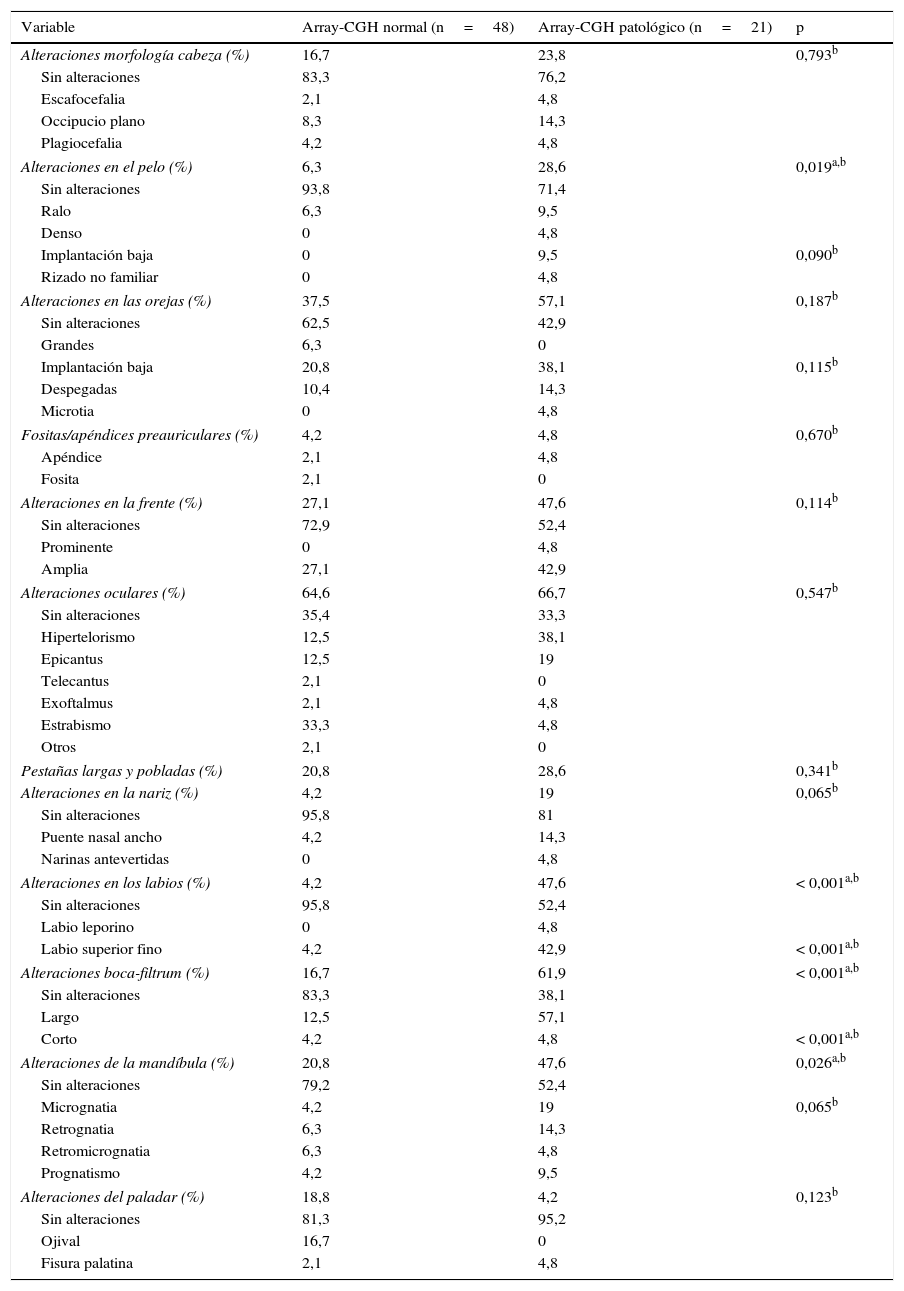
Con respecto al estudio de los rasgos dismórficos cráneo-faciales se realizaron 2 comparaciones: presencia o ausencia de cualquier tipo de alteración en cada categoría (ojos, orejas, mandíbula, etc.) mediante test de Fisher y la comparación de los tipos de alteración encontradas en cada categoría mediante chi al cuadrado (tabla 3).
Comparación de los rasgos dismórficos cráneo-faciales
| Variable | Array-CGH normal (n=48) | Array-CGH patológico (n=21) | p |
|---|---|---|---|
| Alteraciones morfología cabeza (%) | 16,7 | 23,8 | 0,793b |
| Sin alteraciones | 83,3 | 76,2 | |
| Escafocefalia | 2,1 | 4,8 | |
| Occipucio plano | 8,3 | 14,3 | |
| Plagiocefalia | 4,2 | 4,8 | |
| Alteraciones en el pelo (%) | 6,3 | 28,6 | 0,019a,b |
| Sin alteraciones | 93,8 | 71,4 | |
| Ralo | 6,3 | 9,5 | |
| Denso | 0 | 4,8 | |
| Implantación baja | 0 | 9,5 | 0,090b |
| Rizado no familiar | 0 | 4,8 | |
| Alteraciones en las orejas (%) | 37,5 | 57,1 | 0,187b |
| Sin alteraciones | 62,5 | 42,9 | |
| Grandes | 6,3 | 0 | |
| Implantación baja | 20,8 | 38,1 | 0,115b |
| Despegadas | 10,4 | 14,3 | |
| Microtia | 0 | 4,8 | |
| Fositas/apéndices preauriculares (%) | 4,2 | 4,8 | 0,670b |
| Apéndice | 2,1 | 4,8 | |
| Fosita | 2,1 | 0 | |
| Alteraciones en la frente (%) | 27,1 | 47,6 | 0,114b |
| Sin alteraciones | 72,9 | 52,4 | |
| Prominente | 0 | 4,8 | |
| Amplia | 27,1 | 42,9 | |
| Alteraciones oculares (%) | 64,6 | 66,7 | 0,547b |
| Sin alteraciones | 35,4 | 33,3 | |
| Hipertelorismo | 12,5 | 38,1 | |
| Epicantus | 12,5 | 19 | |
| Telecantus | 2,1 | 0 | |
| Exoftalmus | 2,1 | 4,8 | |
| Estrabismo | 33,3 | 4,8 | |
| Otros | 2,1 | 0 | |
| Pestañas largas y pobladas (%) | 20,8 | 28,6 | 0,341b |
| Alteraciones en la nariz (%) | 4,2 | 19 | 0,065b |
| Sin alteraciones | 95,8 | 81 | |
| Puente nasal ancho | 4,2 | 14,3 | |
| Narinas antevertidas | 0 | 4,8 | |
| Alteraciones en los labios (%) | 4,2 | 47,6 | < 0,001a,b |
| Sin alteraciones | 95,8 | 52,4 | |
| Labio leporino | 0 | 4,8 | |
| Labio superior fino | 4,2 | 42,9 | < 0,001a,b |
| Alteraciones boca-filtrum (%) | 16,7 | 61,9 | < 0,001a,b |
| Sin alteraciones | 83,3 | 38,1 | |
| Largo | 12,5 | 57,1 | |
| Corto | 4,2 | 4,8 | < 0,001a,b |
| Alteraciones de la mandíbula (%) | 20,8 | 47,6 | 0,026a,b |
| Sin alteraciones | 79,2 | 52,4 | |
| Micrognatia | 4,2 | 19 | 0,065b |
| Retrognatia | 6,3 | 14,3 | |
| Retromicrognatia | 6,3 | 4,8 | |
| Prognatismo | 4,2 | 9,5 | |
| Alteraciones del paladar (%) | 18,8 | 4,2 | 0,123b |
| Sin alteraciones | 81,3 | 95,2 | |
| Ojival | 16,7 | 0 | |
| Fisura palatina | 2,1 | 4,8 | |
A todos los niveles se encontró una mayor proporción de rasgos dismórficos en los individuos que presentaron un test genético patológico con excepción del paladar. De forma específica, las partes en las que se encontraron alteraciones con una mayor prevalencia en el grupo con el test array-CGH alterado con nivel significativo (p < 0,05) fueron las siguientes: el pelo (p = 0,019) con una mayor proporción de pelo mal implantado (OR: 6; IC del 95%: 1,33-26,9), los labios (p < 0,001), con una mayor proporción de labio superior fino (p < 0,001) (OR: 20,6; IC del 95%, 3,99-109,37); la boca-filtrum (p < 0,001) (OR: 8,13; IC del 95%: 2,54-25,9), destacando la presencia de filtrum largo (p < 0,001) y la mandíbula (p = 0,026) (OR: 3,45; IC del 95%, 1,15-10,42), siendo la micrognatia la característica más frecuentemente encontrada rozando la significación estadística (p = 0,065) (tabla 3). El 68,4% de los individuos con un array-CGH patológico presentan más de 3 rasgos dismórficos frente al 31,2% de los individuos con un resultado normal con un nivel de significación inferior a 0,001 (OR: 20,9; IC del 95%: 4,31-101,4).
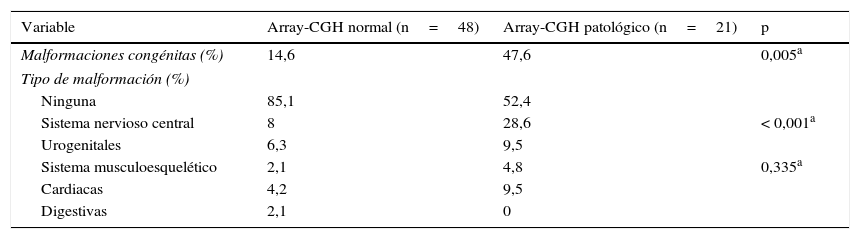
La presencia de malformaciones congénitas fue mayor en el grupo que presentó un resultado del test de array-CGH patológico a nivel general (p = 0,005) y en los distintos subgrupos, excepto a nivel digestivo. El tipo de malformación congénita más frecuentemente encontrada en la muestra analizada fue a nivel del sistema nervioso central en ambos grupos (donde también se han incluido las alteraciones medulares), seguido de las cardiacas y urogenitales pero en este caso sin diferencias significativas entre las 2s cohortes (tabla 4).
Presencia de malformaciones congénitas
| Variable | Array-CGH normal (n=48) | Array-CGH patológico (n=21) | p |
|---|---|---|---|
| Malformaciones congénitas (%) | 14,6 | 47,6 | 0,005a |
| Tipo de malformación (%) | |||
| Ninguna | 85,1 | 52,4 | |
| Sistema nervioso central | 8 | 28,6 | < 0,001a |
| Urogenitales | 6,3 | 9,5 | |
| Sistema musculoesquelético | 2,1 | 4,8 | 0,335a |
| Cardiacas | 4,2 | 9,5 | |
| Digestivas | 2,1 | 0 | |
Análisis estadístico.
En la tabla 5 se muestran los principales rasgos dismórficos, la presencia de malformaciones congénitas y los antecedentes familiares de DI de cada uno de los casos con resultado patológico.
Principales características de los individuos con resultado de array-CGH patológico
| Caso | Alteración genética | HFDI | Fenotipo | Malformación |
| 1 | Del: 7p22.3p22.1 Dup: 15q26.3 (Trans) | + | Bajo peso, micro y dolicocefalia. Frente prominente, hipertelorismo, filtrum largo, retrognatia. Hexadactilia en ambos pies | Cardiaca |
| 2 | Dup: 7p22.3p22.1 Del: 15q26.3 (Trans) | + | Occipucio plano, pelo ralo, orejas de implantación baja, labios finos, filtrum largo, micrognatia. Hiperlaxitud articular | Pie zambo |
| 3 | Del: 5q12.1q13.2 | – | Microcefalia, frente amplia, sinefridia, implantación baja pelo, pestañas largas, retrognatia, filtrum largo. Macroglosia. Tórax excavatum | – |
| 4 | Del: 2q31.2q32.3 Dup: Xq24 | – | Macrocefalia. Frente amplia, hipertelorismo, epicantus, orejas de implantación baja, retrognatia, filtrum largo, labio superior fino | Cardiaca/SNC |
| 5 | Dup: 17q12 | + | Hipertelorismo, pestañas largas, labio superior fino, filtrum largo | – |
| 6 | Del: 7q33q36.1 | – | Epicantus interno y puente nasal ancho | Fisura palatina |
| 7 | Del: 15q21.1q21.2 Marfan like | – | Hábito longilíneo. Epicantus interno, pabellones auriculares despegados, labio superior fino. Escoliosis. Hiperlaxitud articular | Urogenital |
| 8 | Del: 2q21.1q21.31 | – | Micro y dolicocefalia. Orejas de implantación baja, frente amplia, hipertelorismo, pestañas largas, labio fino, filtrum largo | Urogenital |
| 9 | Del: 1q21.1 | – | – | – |
| 10 | Del:3q26.31 | – | Pabellones auriculares despegados, epicantus, puente nasal ancho, filtrum largo | – |
| 11 | Del: 15q11.2q13.1 S. Angelman | – | Fallo medro, microcefalia. Pelo rubio ralo. Piel y ojos muy claros. Frente amplia, micrognatia | – |
| 12 | Del: 16p11.2 | + | Talla alta. Frente amplia, hipertelorismo, pestañas largas | Microtia |
| 13 | Del: 9q34.11 | – | Orejas de implantación baja, hipertelorismo, filtrum largo, micrognatia, clinodactilia en ambas manos. Hipertricosis | SNC |
| 14 | Del: 14q23.1q23.2 | – | Micro y plagiocefalia, orejas de implantación baja, frente amplia, epicantus, labios finos, filtrum corto | SNC |
| 15 | Del: 1q21.1 S. X frágil | – | Microcefalia, frente amplia, pabellones auriculares despegados, hipertelorismo, filtrum largo, retromicrognatia | – |
| 16 | Del: 17p11.2 S. Smith-Magenis | – | Conductas agresivas, trastorno del sueño. Orejas de implantación baja, hipertelorismo, prognatismo. Braquidactilia. Malposición dental | SNC |
| 17 | Del: 15q13.2q13.3 | – | – | – |
| 18 | Del: 8p22p21.1 Dup: 8p12 | – | Occipucio plano, orejas de implantación baja, hipertelorismo, narinas antevertidas, filtrum largo. Tórax en escudo | SNC |
| 19 | Dup: 22q11.21 | – | – | – |
| 20 | Dup: Xp22.2 | – | Fallo medro, microcefalia, Frente amplia, labio superior fino con filtrum largo. Tetraparesia espástico distónica | SNC |
| 21 | Del: 12q21.2 | – | Frente amplia, hipoplasia malar, paladar ojival | – |
| 22 | Dup: 15q11.2 | – | – | SNC |
Del: deleción; Dup: duplicación; HFDI: historia familiar de discapacidad intelectual; Trans: translocación; SNC: sistema nervioso central.
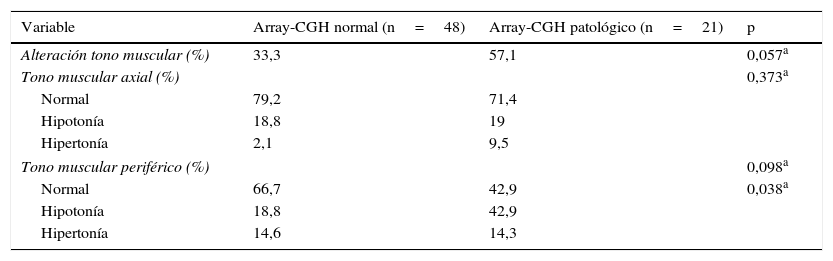
En el estudio del tono muscular se detectó un predominio de la hipotonía sobre la hipertonía tanto a nivel axial como periférico, con una distribución similar en ambos grupos de análisis de la hipotonía axial, mientras que la proporción de pacientes con hipotonía periférica fue mayor en el grupo con resultado array-CGH patológico (tabla 6).
Alteraciones del tono muscular axial y periférico
| Variable | Array-CGH normal (n=48) | Array-CGH patológico (n=21) | p |
|---|---|---|---|
| Alteración tono muscular (%) | 33,3 | 57,1 | 0,057a |
| Tono muscular axial (%) | 0,373a | ||
| Normal | 79,2 | 71,4 | |
| Hipotonía | 18,8 | 19 | |
| Hipertonía | 2,1 | 9,5 | |
| Tono muscular periférico (%) | 0,098a | ||
| Normal | 66,7 | 42,9 | 0,038a |
| Hipotonía | 18,8 | 42,9 | |
| Hipertonía | 14,6 | 14,3 | |
Análisis estadístico.
La realización del estudio de regresión logística multivariante permitió identificar los principales factores asociados a presentar un resultado alterado de forma independiente, que fueron la presencia de antecedentes familiares de DI, más de 3 rasgos dismórficos cráneo-faciales y alteraciones a nivel de los labios.
Las pruebas genéticas que se habían realizado previamente en el grupo del array-CGH patológico no fueron concluyentes. Se llevó a cabo cariotipo en 15 individuos (4 de ellos de alta resolución) y estudio de deleciones subteloméricas en 5 casos. Se descartó síndrome X frágil en 14 varones y 2 mujeres. El caso número 15 merece especial atención, ya que se descartó síndrome X frágil en el año 2006 por la técnica Southern blot. Ante fenotipo compatible (exceptuando la microcefalia), se repitió el estudio en el año 2012, presentando más de 250 repeticiones del trinucleótido CGG mediante técnica PT-PCR.
DiscusiónLa técnica de array-CGH ha supuesto un gran avance en el estudio de la DI/RGD de forma que se propone como primera prueba a realizar en esta patología por gran parte de los autores13,14.
A pesar de la limitación del tamaño muestral, es importante señalar que se ha realizado el estudio pormenorizado de las características de forma ciega (sin conocer el resultado del array-CGH), por un único investigador, lo que disminuye la variabilidad y aumenta la validez interna.
El rendimiento encontrado en nuestra muestra ha sido superior a la mayoría de las series de datos publicados que varían entre el 5 y el 25%13,14. Este hallazgo puede ser debido a los criterios de selección que se siguieron inicialmente a la hora de realizar el test genético en los que se tendió a elegir a aquellos pacientes con antecedentes familiares de DI, rasgos dismórficos y/o malformaciones congénitas. En aquellas series en las que se usaron criterios semejantes se han obtenido índices de detección similares8,15.
Un 60% de las alteraciones encontradas fueron deleciones. Este hecho puede tener una doble explicación, tanto técnica como biológica. Técnicamente, existe mayor probabilidad que las duplicaciones no sean detectadas. Biológicamente, se ha descrito que las duplicaciones causan fenotipos menos severos y por ello se han podido perder casos en la selección de la muestra. También se ha descrito que la frecuencia de que se produzcan duplicaciones al azar en el genoma humano es inferior a la de deleciones14.
La distribución de las CNV parece no ser al azar, existiendo puntos calientes donde se encuentran con mayor frecuencia. Estas zonas suelen ser ricas en genes y regiones con duplicaciones segmetarias16. En nuestra muestra, la mayoría de las CNV afectan a genes implicados en la señalización y la adhesión celular, o bien codifican para proteínas estructurales del sistema nervioso central17.
Con respecto a la herencia, los casos n.° 1 y 2 son secundarios a translocaciones equilibradas maternas. La detección de translocaciones equilibradas crípticas es extraordinariamente importante debido al alto riesgo de recurrencia. Su detección permite el correcto asesoramiento genético a toda la familia. En el caso 5, la deleción es heredada de su padre, que presenta un grado inferior de DI que el probando, lo que nos hace sospechar de la expresividad variable de la alteración. El caso n.° 20 presenta una duplicación a nivel del cromosoma X, heredada de la madre sin fenotipo reportado. Esta disomía contiene genes descritos en OMIM asociados a patología. El hecho de que la madre no presente clínica puede ser debido al fenómeno de inactivación selectiva del cromosoma X. Por último, los casos n.° 19 y 22 son heredados de la madre sana y afectan a autosomas. El caso n.° 19 presenta una microduplicación en el cromosoma 22, que ha sido descrito en la literatura asociado a penetrancia incompleta y expresión muy variable que incluye desde portadores con inteligencia normal hasta portadores con DI severa y un alto número de pacientes que heredan la alteración de un progenitor no afectado18. El caso n.° 22 presenta una duplicación en una zona de microduplicaciones/deleciones frecuentes del cromosoma 15. En la literatura científica existen numerosos casos en los que la duplicación es heredada de un progenitor con fenotipo normal. Se postula que la clínica es más grave cuando se hereda por vía materna, existiendo una gran variabilidad intrafamiliar (desde inteligencia normal hasta alteraciones cognitivas graves acompañadas de epilepsia)19.
En las CNV clasificadas como de significado incierto, las alteraciones han sido heredadas de un progenitor sano en el 90% de los casos. Este hecho aislado debe interpretarse de forma cautelosa, ya que aunque a día de hoy no se haya podido demostrar, estos desequilibrios cromosómicos pueden contribuir al fenotipo del paciente como consecuencia de la expresividad variable de los mismos, efectos epigenéticos o bien debido a una mutación recesiva en el alelo no delecionado14.
Con respecto a la antropometría, en estudios previos se ha detectado una asociación entre la microcefalia y el resultado patológico del array-CGH20. Al calcular la proporción de individuos con microcefalia grave (perímetro cefálico inferior a –3 desviaciones estándar), la proporción es muy superior en el grupo con resultado del array-CGH patológico aunque sin alcanzar significación estadística (tabla 1). Estos hallazgos nos hacen presuponer que si nuestra muestra tuviese un número mayor de pacientes es probable que se reprodujesen los resultados del estudio citado con respecto a la microcefalia. Otros autores han encontrado con mayor frecuencia trastornos del crecimiento en los individuos que presentaban un resultado del array-CGH patológico: Shoukier et al.20 refieren fallo de medro, Roselló et al.8 talla alta, mientras que Vulto-van Silfhout et al.15, como en el presente trabajo, no encuentran asociación entre la evolución de la altura/peso y el resultado del test genético. Esta variabilidad encontrada en la literatura científica con respecto al crecimiento puede ser debida a los criterios de selección de los casos, así como al resultado de la prueba de array, ya que hay síndromes que se acompañan de fallo de medro y otros de sobrecrecimiento. Son necesarios estudios con un mayor número de individuos para determinar la implicación de los desórdenes de crecimiento en las alteraciones estructurales cromosómicas.
Numerosos autores han publicado la asociación entre la presencia de alteraciones cromosómicas y las malformaciones congénitas8,15,20. En la mayoría de las series las más prevalentes fueron las malformaciones cardiacas y en el presente estudio fueron a nivel del sistema nervioso central, principalmente las alteraciones del cuerpo calloso. Este hallazgo coincide con lo publicado por Preiksaitiene et al.21. Al revisar la metodología de los diferentes estudios, se pone de manifiesto que los criterios de selección influyen en el tipo de malformación más frecuente. No obstante, en nuestro estudio la distribución de las malformaciones fue absolutamente aleatoria.
La presencia de rasgos dismórficos constituye una variable muy importante en nuestra muestra debido a la alta prevalencia de los mismos en los individuos afectos de DI/RGD. Como prueba de ello, tras la exploración pormenorizada de los mismos, solamente en un 25% de los individuos con array-CGH normal y en un 13,6% de los casos que presentan CNV patológicas no se detectó ninguna alteración morfológica menor. En nuestro estudio, el pelo mal implantado, el filtrum largo, el labio superior fino y la micrognatia fueron las características en las que se encontraron diferencias significativas. Aunque el tipo de rasgo dismórfico más frecuente varía mucho entre unas series y otras, la presencia de varios rasgos en conjunto, constituye un buen indicador de la posible existencia de un desequilibrio cromosómico15.
Se han publicado varios trabajos en los que se compara el rendimiento de las diferentes técnicas diagnósticas genéticas aplicadas al estudio de la DI. Miller et al.13 realizaron una revisión de 33 estudios que abarcaban 21.698 pacientes y concluyeron que mientras el cariotipo de alta resolución tenía un rendimiento del 3% (excluyendo el síndrome de Down) y la aplicación de FISH a los telómeros un 2,4%, la técnica array-CGH alcanzaba un 12% de diagnósticos en pacientes no seleccionados. En un estudio más reciente realizado en Europa sobre 36.325 pacientes en los que se realizó cariotipo y array-CGH se detectaron CNV patológicas en un 19% de los individuos no seleccionados y solamente un 0,78% de las alteraciones genéticas no hubiesen sido detectadas sin la realización del cariotipo22. Ambos trabajos concluyen que el estudio mediante array debe ser la primera prueba en el abordaje diagnóstico de los pacientes afectados de DI de causa no explicada.
Dada la reciente incorporación del array-CGH a la práctica clínica, todavía hay muchas limitaciones a la hora de llevar a cabo una correcta interpretación de los resultados, así como la consiguiente información que se les da a las familias. En estudios previos reportados se detectó una gran variabilidad en la misma, así como en el manejo posterior de los pacientes entre diferentes profesionales23. Ambos factores pueden mejorarse si clínicos y genetistas trabajan en estrecha colaboración y se diseñan protocolos que disminuyan la variabilidad clínica y aumenten la calidad de la asistencia. Por otra parte, si a medida que la técnica se extiende en la práctica diaria, los resultados obtenidos se comparten y pueden ser consultados a través de bases de datos públicas en conjunción con las características fenotípicas de cada caso, será más sencillo definir la patogenicidad de cada una de las alteraciones encontradas.
Con respecto al coste efectividad de la técnica, recientemente se ha publicado un estudio que concluye que la realización de un cariotipo molecular es más económico que la realización de un cariotipo convencional y las subsiguientes pruebas (FISH aplicado a telómeros, etc.) y además se obtienen entre un 15 y un 20% más de resultados diagnósticos que por la vía tradicional24.
Actualmente, el test genético array-CGH constituye una prueba de primer orden en el estudio de la DI/RGD, ya que tiene un rendimiento superior a las pruebas genéticas convencionales (cariotipo, FISH/MLPA aplicado a telómeros, etc.). Aunque existen factores que se asocian a detectar con mayor probabilidad reordenamientos cromosómicos, estos no tienen una fuerza de discriminación suficiente para preseleccionar los candidatos ideales para realización de esta prueba. En nuestro servicio hemos modificado nuestro protocolo de estudio de la DI/RGD y la técnica array-CGH se ofrece a toda la población de individuos con DI/RGD de etiología no conocida.
FinanciaciónNo se ha recibido financiación.
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.
El presente trabajo no ha sido presentado en la Reunión Anual de la SEN o en otras reuniones.

