



El síndrome de la persona rígida (SPR) es un raro trastorno que se caracteriza por rigidez muscular y espasmos dolorosos intermitentes. Etiológicamente parece existir una base autoinmune por la presencia de anticuerpos actuando sobre el sistema GABAérgico y la asociación con otras enfermedades autoinmunes1–3. Afecta especialmente a la musculatura de tronco y extremidades, y existen desde formas generalizadas a formas focales y otras variantes más raras con afectación encefalomielítica.
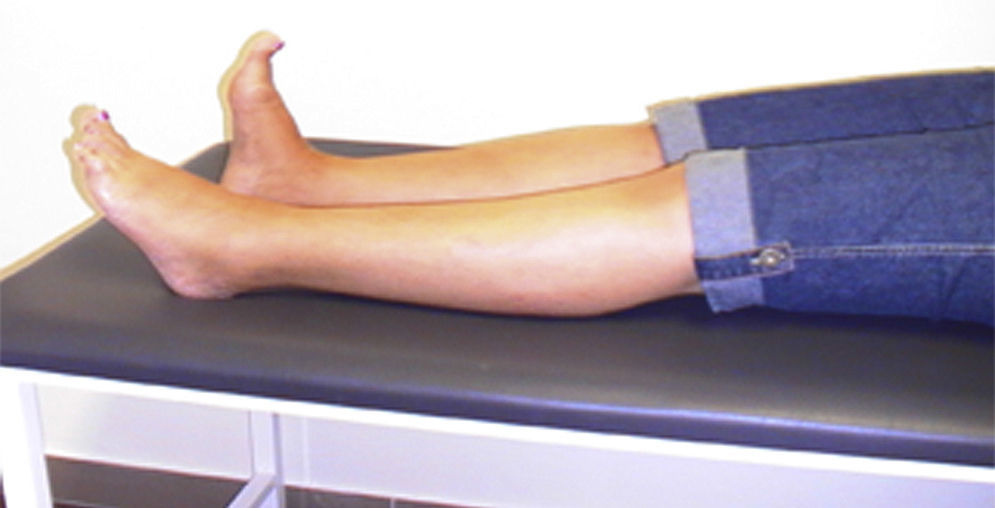
Presentamos el caso de una mujer que a los 46 años inicia espasmos de dolor en pie izquierdo y postura anómala en extensión del primer dedo, evolucionando en 5 años a todo el pie (fig. 1), con síntomas de agarrotamiento en toda la pierna izquierda y caídas frecuentes.

Se realizaron IRM craneal e IRM medular que resultaron normales, y un SPECT con ioflupano I-123, también normal. Los estudios analíticos: análisis general, metabolismo del cobre, inmunología básica, función tiroidea, marcadores tumorales y anticuerpos antineuronales resultaron normales a excepción de: anticuerpos anti-GAD (descarboxilasa del ácido glutámico) >250U/l en 2 determinaciones, ANA positivos a nivel moderado (1/160) (patrón punteado reticular) y anticuerpos antiislotes pancreáticos (ICAS) positivos. Los antianfifisina fueron negativos. Un estudio de cribado de neoplasia oculta fue negativo.
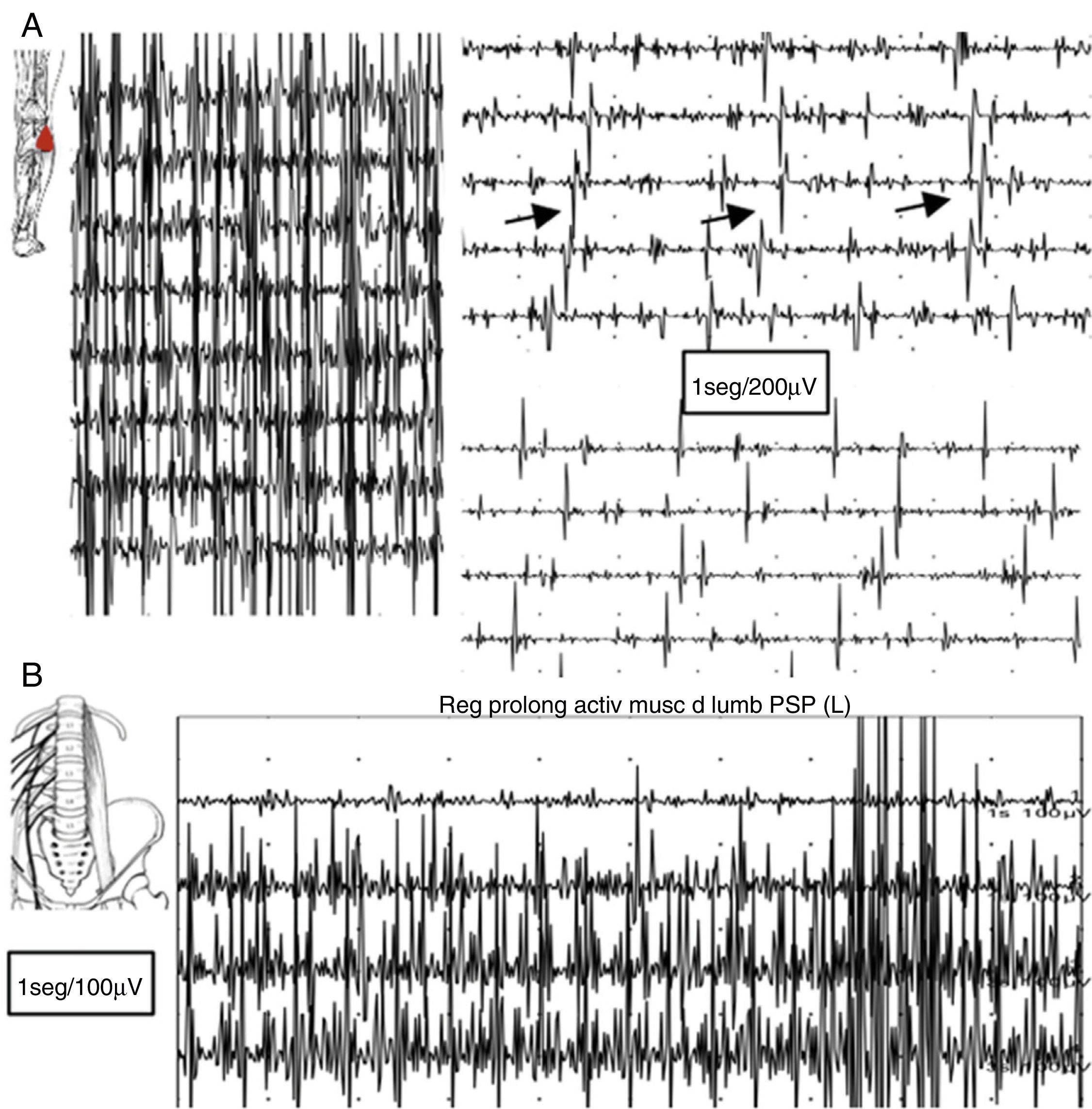
En el estudio electromiográfico (EMG) se evidenció una actividad muscular continua en miembro inferior izquierdo de predominio flexo-extensor distal y en musculatura paravertebral lumbar, con unidades motoras de morfología normal que no desaparecen con maniobras de distracción ni con la activación de antagonistas (fig. 2).
 Registro electromiográfico del músculo gemelo externo: tras contracción voluntaria persiste falta de relajación muscular con breves contracciones intercaladas semirrítmicas aparentemente a 5Hz. B) Registro en reposo mediante electrodo de aguja concéntrico en musculatura paravertebral lumbar: falta de relajación con actividad continua de PUM normales.")
A) Registro electromiográfico del músculo gemelo externo: tras contracción voluntaria persiste falta de relajación muscular con breves contracciones intercaladas semirrítmicas aparentemente a 5Hz. B) Registro en reposo mediante electrodo de aguja concéntrico en musculatura paravertebral lumbar: falta de relajación con actividad continua de PUM normales.
Con base en estos estudios, la paciente fue diagnosticada de SPR en su fenotipo parcial (Stiff-leg syndrome). Desde entonces ha recibido tratamiento con diazepan, baclofeno y gabapentina, mejorando mucho los espasmos y el dolor de pie y pierna izquierdos, aunque sus caídas han seguido un curso fluctuante, apareciendo además episodios de «bloqueo» de la marcha ante situaciones estresantes. Tras 9 años presenta evolución desfavorable con mayor rigidez de la extremidad, mayor hiperlordosis lumbar y gran dificultad para la deambulación, con bloqueos y alteración en la ignición de la marcha, así como aumento de las caídas, asociando un cuadro de ansiedad, fobias y conductas de evitación.
El SPR se engloba dentro de los síndromes de hiperexcitabilidad con afectación de la neurona motora espinal. Clínicamente se caracteriza por rigidez progresiva de la musculatura, predominantemente axial, con presencia de espasmos superimpuestos e hipersensibilidad a estímulos externos3,4. Existen formas incompletas, subtipos o variantes, tales como: el síndrome de la extremidad rígida (como es nuestro caso) —puede evolucionar a una forma completa—; el SPR asociado a mioclonías; el SPR asociado a epilepsia y distonía; el SPR con manifestaciones neurooftalmológicas; el SPR con rigidez progresiva y encefalomielitis o la variante cerebelosa.
Los espasmos episódicos se desencadenan ante factores como el estrés emocional, estímulos externos súbitos, frío, infecciones intercurrentes o movimientos rápidos, asociando típicamente manifestaciones psiquiátricas como fobia a salir de casa y una depresión reactiva que puede confundir y dificultar el diagnóstico. Se ha postulado que factores estresantes psicológicos/psicosociales pueden activar un proceso inmune oculto5.
Los hallazgos de EMG se caracterizan por una actividad continua de unidades motoras en reposo persistente a pesar de los intentos de relajación, con una morfología normal y sin evidenciar signos de denervación aguda ni actividad espontánea y con un patrón de reclutamiento normal3,6,7. Se afecta simultáneamente musculatura agonista y antagonista, y se producen los llamados espasmos reflejos inducidos.
Fisiopatológicamente se basa en la presencia de anticuerpos anti-GAD que actúan sobre el sistema GABAérgico inhibiendo su función y provocando una hiperexcitabilidad muscular anormal, aunque no todos los pacientes descritos presentan este anticuerpo (60-90% de los casos)2,3, existiendo otras enfermedades relacionadas con su presencia8.
Actualmente, los tratamientos disponibles se basan en aumentar la actividad del GABA (benzodiacepinas, baclofeno) y el tratamiento inmunomodulador y/o inmunosupresor (prednisona, inmunoglobulinas iv, plasmaféresis, rituximab, micofenolato, ciclofosfamida, azatioprina o metotrexato)9. Se han ensayado también fármacos antiepilépticos (valproato, vigabratrina, gabapentina, levetiracetam, tiagabina) con resultados dispares. También se ha probado tratamiento con onabotulinumtoxin A, aunque la eficacia no está todavía demostrada9. Además, se recomienda el tratamiento precoz de los síntomas psiquiátricos que se desarrollan a lo largo de la evolución de la enfermedad5.
Consideramos importante tener en cuenta esta entidad en el diagnóstico diferencial de las distonías focales especialmente de extremidad inferior10, dada la marcada incapacidad que producen, que puede ser mejorada cuando es correctamente diagnosticada y tratada.
Conflicto de interesesLos autores de este manuscrito declaran la ausencia de conflicto de intereses.

