



Hirayama disease is a rare cervical myelopathy, predominantly affecting young men, and which presents with distal atrophy of the upper limbs as its first and main symptom. It must be differentiated from motor neuron diseases because its natural history is different and because HD tends to stabilise in less than 5 years.
Diagnosis is based on clinical findings and dynamic flexion MRI showing segmental spinal muscular atrophy, detachment of the posterior dura mater and venous congestion in the epidural space.
The tendency is to indicate conservative treatment and no indications for surgery have been established.
PatientsWe present 4 cases meeting both clinical criteria and dynamic MRI imaging criteria for a diagnosis of Hirayama disease. Two have stabilised spontaneously over the course of many years, and MRI scans show that typical changes have disappeared. Another case also remains stable following a shorter observation time. The fourth case is a young man who developed severe myelopathy in just over a year, and therefore underwent surgery. While his follow-up time is still short, his condition remains stable.
ConclusionsOur 4 cases suggest that the condition of most patients with Hirayama stabilises naturally; patients should be evaluated for surgery on an individual basis, and surgery should probably be limited to the most severe cases that have progressed quickly.
La enfermedad de Hirayama es una rara mielopatía cervical mucho más frecuente en hombres jóvenes, que cursa con atrofia distal en los brazos como primer y principal síntoma. Es importante diferenciarla de las enfermedades de las motoneuronas porque su historia natural es distinta, con tendencia hacia la estabilización en menos de 5 años.
El diagnóstico se basa en los hallazgos clínicos y en la RM dinámica (en flexión del cuello), que detecta la atrofia medular segmentaria, el despegamiento posterior de la dura madre y la congestión venosa en el espacio epidural.
La tendencia es a indicar un tratamiento conservador pero no está establecido cuál puede ser el papel de la cirugía.
PacientesSe presentan 4 casos que cumplen los criterios clínicos y de imagen en RM dinámica para diagnóstico de enfermedad de Hirayama. Dos están en fase de estabilización espontánea después de muchos años de evolución y en la RM se demuestra la desaparición de las alteraciones típicas. Otro caso también permanece estable con menos tiempo de observación. El cuarto caso corresponde a un joven que en poco más de un año presenta una mielopatía grave, por lo que ha sido operado, manteniéndose estable tras una observación todavía corta.
ConclusionesNuestros 4 casos indican que la mayoría de los pacientes con enfermedad de Hirayama se estabilizan de manera natural y que la intervención quirúrgica debe ser una decisión individual, probablemente limitada a los casos más graves con una evolución muy breve.
Hirayama disease was first defined in Japanese in 1959, and in English in 1963,1 as unilateral focal amyotrophy of one of the upper limbs; its course differed from that of motor neuron degenerative diseases. According to the first published autopsy report,2 the cervical spinal cord lesions were not typical of a motor neuron disease, but rather suggested ischaemic necrosis in the anterior horns of the spinal cord. In subsequent years, his vast experience led Hirayama to describe the disease's clinical and radiological characteristics. This author also suggested the main hypothesis for its pathogenesis (myelopathy secondary to neck flexion) and proposed using a cervical collar as a preventive measure.3
The disease usually presents in young individuals and is much more common among male patients (male-to-female ratio of 20:1), as well as in Asian countries (333 cases were diagnosed in Japan between 1996 and 1998). Contrary to conclusions drawn from the earliest observations, amyotrophy is not always monomelic; instead, it tends to be bilateral, although it is frequently asymmetrical. Fasciculations are infrequent. Atrophy usually affects the muscles in myotomes C7 and C8, and therefore the hands and forearms. The brachioradialis typically shows at least partial preservation, whereas muscles from the myotome C5 are completely preserved. Atrophy rarely affects myotome T1. Patients may present muscle reflexes in the arms despite amyotrophy. No relevant pyramidal signs or sensory alterations are associated. In many patients, cold temperatures worsen hand muscle paresis.
In addition to these basic clinical signs, other more atypical findings have been reported,4,5 including autonomic alterations, fasciculations, minipolymyoclonus, amyotrophy extending to other segments (especially T1), abolished reflexes in the upper limbs, and pyramidal signs in the lower limbs. These signs are considered indicative of severe forms of the disease.6
The most widely accepted hypothesis on the aetiopathogenesis of Hirayama disease postulates that the disease may arise from discrepancies between the growth of the spinal column, spinal cord, and dura mater.3 As MR images show, dura mater detaches from the posterior lamina and displaces the spinal cord, which is compressed and bent against the vertebral column during neck flexion. Spinal cord segments where kyphosis is present are atrophic and frequently appear as hyperintensities, indicating spinal cord lesion. The epidural space contains dilated veins and tissues that display contrast uptake. All these findings are highly specific for a diagnosis of the disease. Some patients may not present the typical signs of flexion myelopathy seen on MR images.7 However, as Hirayama himself points out,3 these cases are rare and may be related to the stage of disease progression in which the MRI scan is performed, since these findings tend to disappear in the chronic phase.8
Surgical treatment is controversial for several reasons: the disease tends to stabilise naturally after 2-3 years of progression, few patients undergo surgery, different surgical techniques can be used, and outcomes are uncertain.4,8,9 With a view to discussing the suitability of surgical treatment, we present 4 patients with Hirayama disease, including a particularly severe case that required surgery.
PatientsPatient 1: 19-year-old malePersonal historyAt the age of 2, this patient underwent successful surgical treatment for aortic coarctation. He plays sports regularly and reports no cardiovascular symptoms. Prior to our department's assessment, a CT angiography of the supra-aortic trunks and an MRI scan of the brachial plexus had yielded normal results.
Medical historyThe patient reported loss of strength and muscle mass in the ulnar region of the right hand in the preceding 2 years. He initially experienced poor motor function in the fourth and fifth digits, especially on cold days. Weakness progressed during the first year and affected finger and hand flexion. The patient also reported atrophy of the hypothenar eminence, interosseous muscles, and medial side of the forearm. However, symptoms had stabilised in the preceding 9 months. The patient reported no pain, paraesthesia, or other sensory alterations.
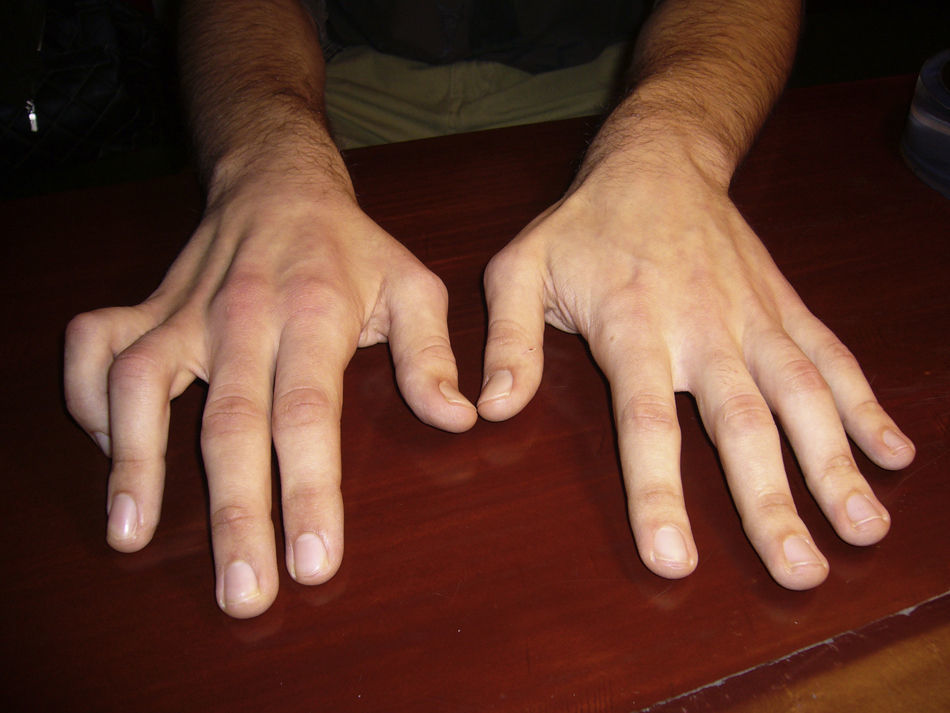
Physical examinationExamination revealed severe amyotrophy of the dorsal interosseous muscles of the right hand and muscle atrophy in the first, third, and fourth interosseous spaces (Fig. 1). The patient exhibited weakness in the abductor pollicis as well as mild weakness of the long flexors of the thumb and index fingers, but no fasciculations or sensory impairment. He presented preserved muscle reflexes and no Babinski sign. Results from the neurological examination were otherwise normal.
Complementary tests
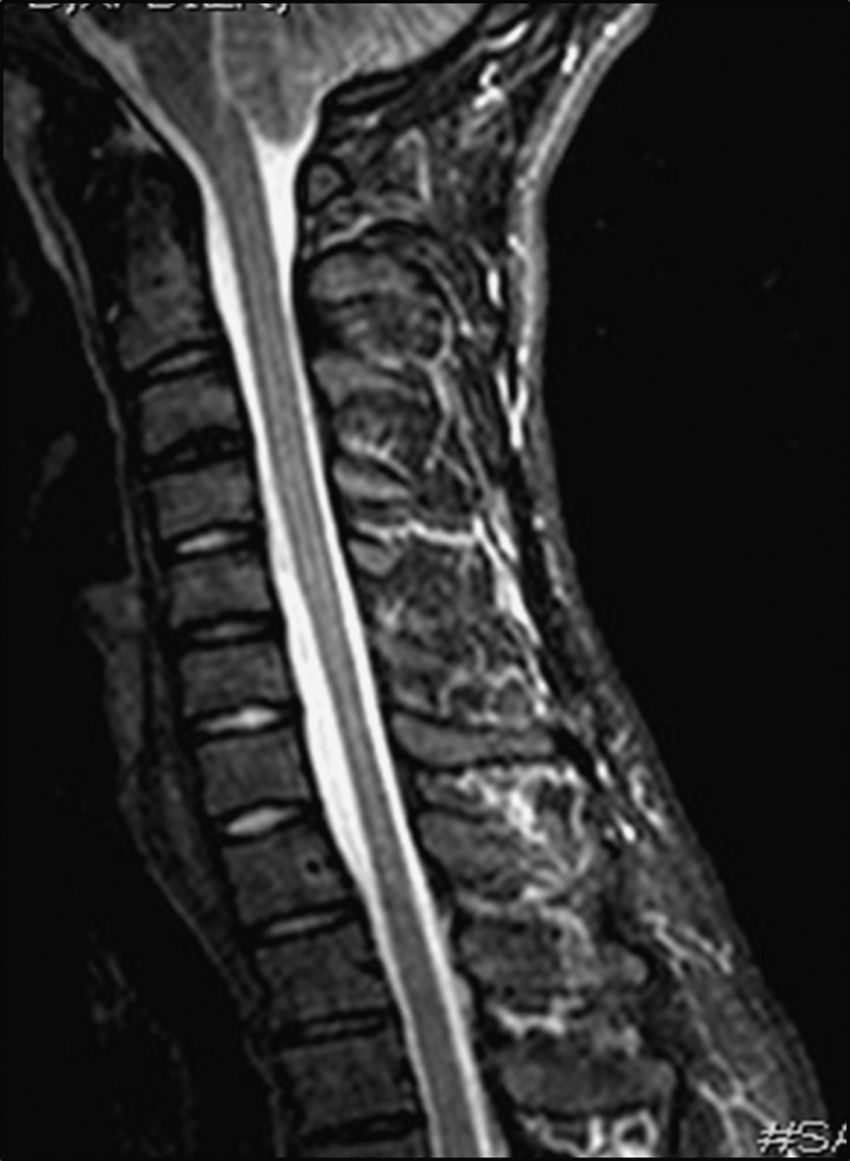
Results from an electromyography study (EMG) performed in March 2009 suggested severe C8 radiculopathy and active denervation. The electroneurography (ENG) study yielded normal results. A cervical MRI scan in the neutral position showed spinal cord thinning at C6 and C7, with no intramedullary signal intensity changes and slight dilation of the subarachnoid spaces at the same level (Fig. 2). MRI scan with neck flexion revealed that dura mater had migrated a considerable distance from the posterior lamina (up to 7mm). There was also engorgement of the posterior epidural venous plexus and anterior displacement of the spinal cord which was touching the posterior wall of the cervical and superior thoracic vertebral bodies (Fig. 3). The MR image also displayed decreased spinal cord diameter in that segment and a slight increase in signal intensity.
 displaying slight spinal cord thinning at C6-C7 and moderate increase of the anterior subarachnoid space.")
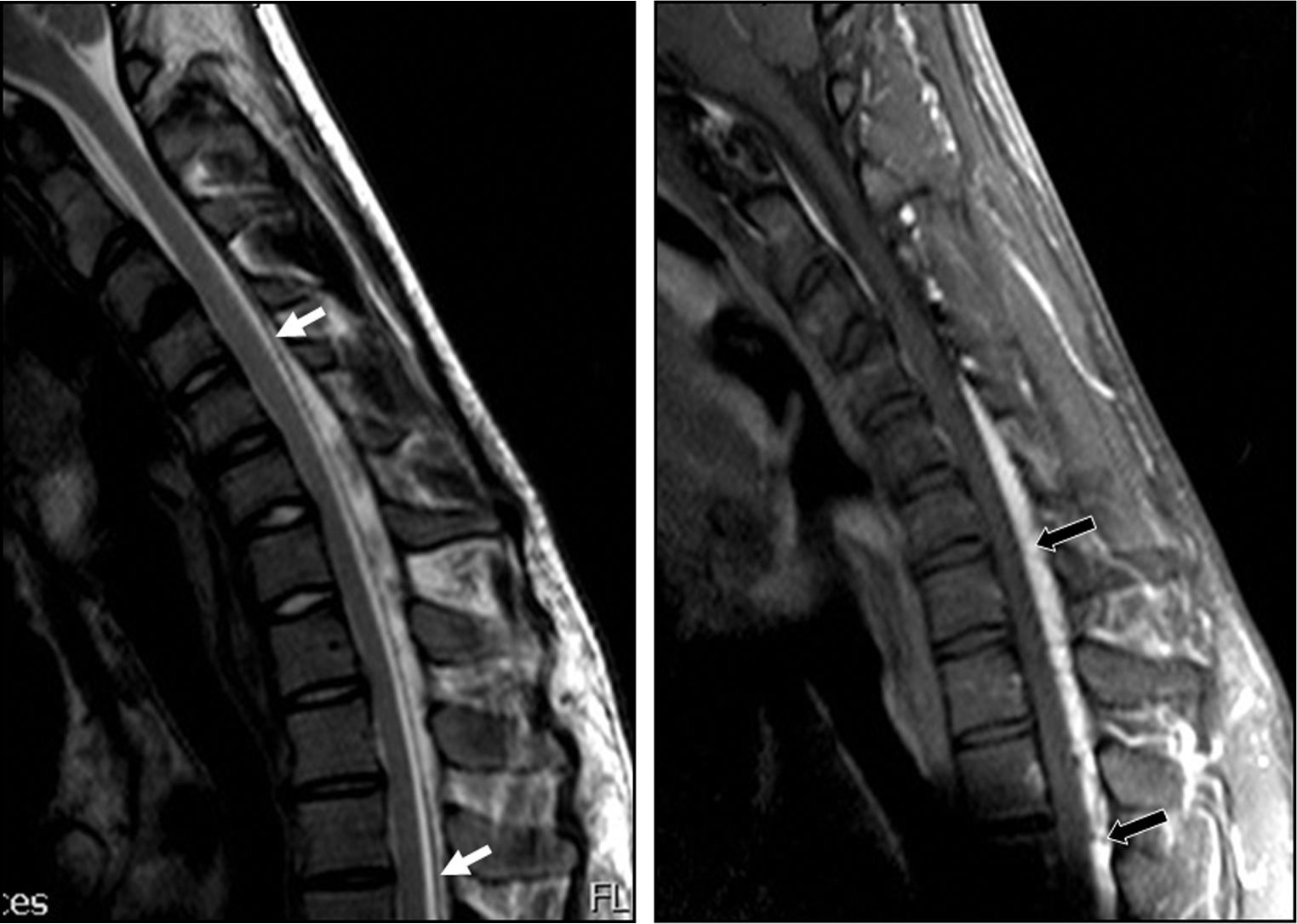
 showing marked dura mater shifting from the posterior lamina (white arrows) and accumulation of liquid and engorgement of the posterior epidural venous plexus (black arrows). The spinal cord has moved forward and shows an angle at C6-C7.")
Patient 1. Flexion cervical MRI scan (T2 and T1) showing marked dura mater shifting from the posterior lamina (white arrows) and accumulation of liquid and engorgement of the posterior epidural venous plexus (black arrows). The spinal cord has moved forward and shows an angle at C6-C7.
After a year and a half of follow-up, the patient presents no signs of progression in either physical examinations or electromyography studies.
Patient 2: 40-year-old male with anxietyPersonal historyThe patient had no relevant clinical history, although he suffered from anxiety and was a hypochondriac. He exercised regularly (racquetball).
Medical historyThe patient was seen for anxiety due to personal problems. The examination revealed predominantly right-sided but bilateral amyotrophy in the hands. The patient explained that he had noticed loss of strength in both hands at least 20 years before. He consulted a doctor but was not given a definitive diagnosis at that time, and symptoms stabilised. He had led a normal life ever since and was even able to play racquetball regularly. He reported no pain or sensory disorders.
Physical examinationThe neurological examination revealed atrophy of the intrinsic musculature of the right hand, palmar muscles, and carpal flexor muscles. The patient presented preserved symmetrical reflexes. No fasciculations or sensory alterations were observed, and no Babinski sign was present. Other results from the examination were normal.
Complementary testsThe EMG showed no spontaneous electrical activity in any muscles. The flexor carpi radialis, triceps, and first dorsal interosseous muscle of the right arm showed a neurogenic pattern. The EMG study also revealed an intermediate pattern in the flexor carpi radialis, triceps, and first dorsal interosseous muscle of the left hand. The ENG study indicated normal conduction velocities in the median and ulnar nerves. The amplitude of the evoked potential of the abductor digiti minimi was 12.5mV. We concluded that there were old neurogenic and partially reinnervated lesions which were severe on the right (C7-C8 level) and mild on the left. The last cervical MR image was performed with the patient's neck flexed and reveals segmental spinal cord atrophy at the mentioned level and dilated spinal canal. No other pathological findings were observed.
ProgressionThe patient's condition has remained stable since the initial consultation.
Patient 3: 27-year-old malePersonal historyNo relevant history.
Medical historyThe patient started noticing tremor in the fingers at the age of 19. Tremor was predominantly postural and affected precise movements. His hands were stiff after prolonged periods of muscle contraction (for example, writing or carrying a bag). He occasionally experienced fasciculations in the first interosseous space of the right hand and loss of strength when he tapped his thumb and index finger together.
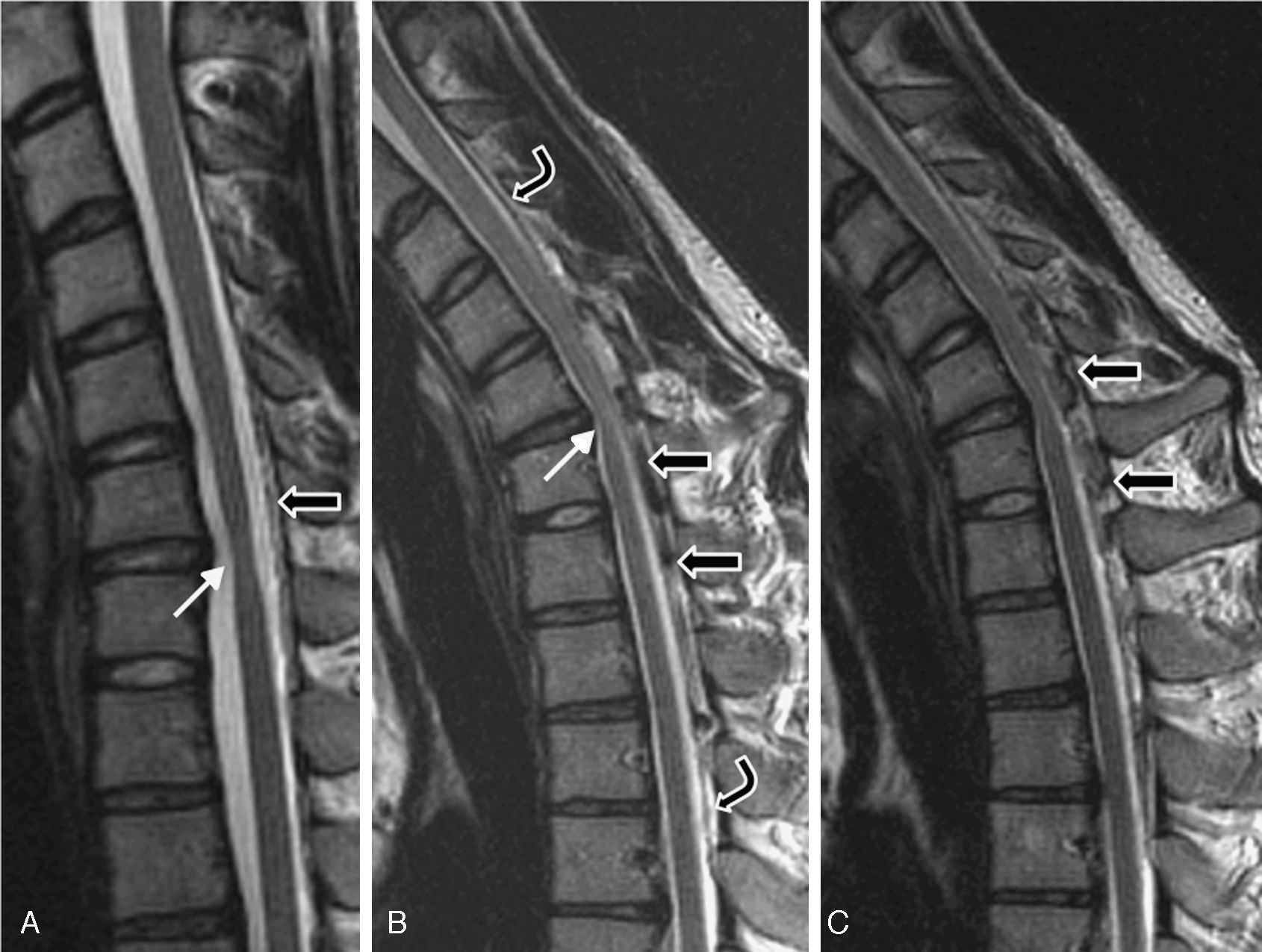
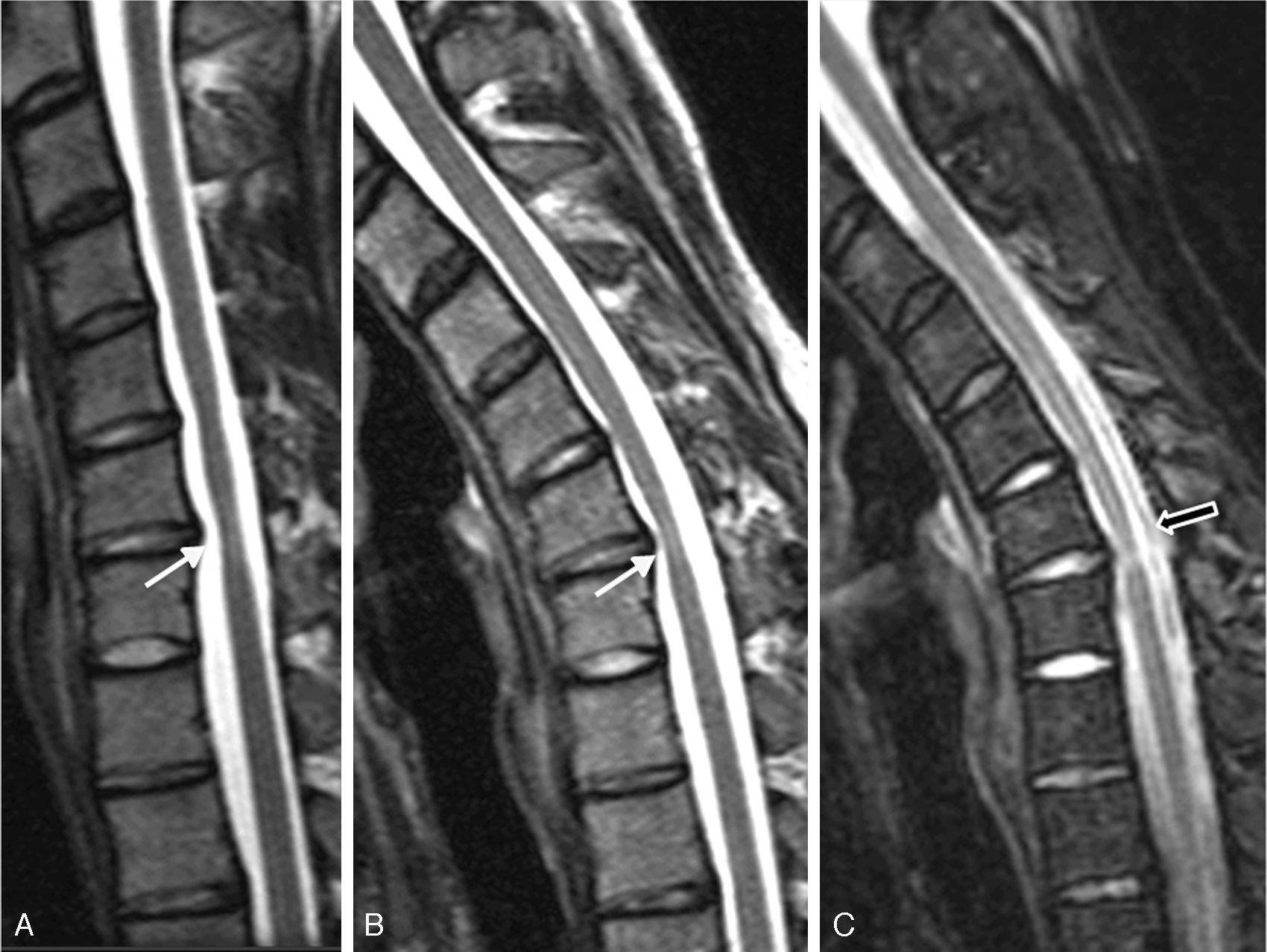
At the age of 20 (in 2004), he was examined in another centre and diagnosed with distal amyotrophy in both arms, but more pronounced on the right side. The EMG revealed fibrillations, positive slow waves, fasciculations with high-amplitude, long-duration potentials, and decreased recruitment in the abductor digiti minimi muscle, the abductor brevis pollicis, the opponens pollicis, and the first dorsal interosseous muscle of the right arm. It also showed chronic neurogenic signs in muscles at levels C8 and T1 that were more intense on the right side. Results from an EMG were normal for all 4 limbs. A cervical MRI detected segmental spinal cord atrophy in the C6-C7 intervertebral area; with neck flexion, it showed thickening of the posterior peridural space, prominent veins, and spinal cord signal changes in the atrophic segment (Fig. 4).
. (A) Neutral position: moderate spinal cord atrophy at disc C6-C7 (white arrow) and minimal posterior detachment of the dura mater. (B) and (C) On flexion: anterior spinal cord compression at site of flexion (white arrow) and faint spinal cord hyperintensity; dura mater detachment (between curved arrows); accumulation of liquids in the epidural space, with signal voids suggestive of venous dilation (black arrows).")
Patient 3. MRI scan performed in 2005 (T2). (A) Neutral position: moderate spinal cord atrophy at disc C6-C7 (white arrow) and minimal posterior detachment of the dura mater. (B) and (C) On flexion: anterior spinal cord compression at site of flexion (white arrow) and faint spinal cord hyperintensity; dura mater detachment (between curved arrows); accumulation of liquids in the epidural space, with signal voids suggestive of venous dilation (black arrows).
Surgical treatment was ruled out and the patient's condition stabilised. In 2011, he visited our centre for a follow-up consultation.
Physical examinationThe examination in 2011 revealed moderate postural tremor (6-8cycles/second). The patient presented marked amyotrophy in the dorsal interosseous muscles of both hands, especially the right hand, with distal amyotrophy of the forearm that predominated on the right side and in the muscles of the palm. Pronator and supinator reflexes were abolished, but biceps and triceps reflexes were normal. The patient presented no Babinski sign and no balance or gait alterations. The rest of the neurological examination yielded normal results.
Complementary testsAn MRI scan performed at our hospital (2011) displays persistence of moderate segmental atrophy of the spinal cord at the C5-C6 disc with an area of mild intramedullary hyperintensity (Fig. 5). On neck flexion, however, dura mater was shown to be only slightly detached from the posterior lamina; no gadolinium uptake or signal voids were seen.
Outcome Neutral position (T2): moderate segmental atrophy persists, (B) (T2), and (C) (STIR), flexion position: minimal dura mater detachment with no signs of occupation of the epidural space.")
A follow-up appointment in 2012 found that the patient remained stable.
Patient 4: 19-year-old malePersonal historyStudent reporting moderate cannabis use and no other relevant history.
Medical historyIn the previous year and a half, the patient had been experiencing progressive weakness in the right hand which was now extending to the arm. He also reported poor motor function in the right leg when walking, which was first noticed by his family a year before. He reported no sensory alterations and no loss of sphincter control, dysphonia, or dysphagia.
Physical examinationMental state, language function, and all cranial nerves were normal. The patient presented general amyotrophy in the right hand and forearm with relative preservation of the brachioradialis. The right triceps was hypotonic, atrophic, weak (strength rating 3-4/5), and presented intense fasciculations, whereas the left triceps was only slightly weak. The right pectoral muscle was clearly atrophic but the left was normal. Biceps, deltoid muscles, and shoulder external rotators were preserved. He displayed normal biceps reflexes, abolished right-sided triceps reflexes, and lively brachioradialis, pronator, and wrist and finger flexor reflexes, especially on the right side, which showed a clonus during the test for the Hoffmann reflex. The right leg presented pyramidal weakness. Patellar and Achilles reflexes were lively in both legs, especially on the right side, where they were clearly pathological and showed ankle clonus. The patient displayed no plantar reflexes except for right-sided withdrawal reflex in response to nociceptive stimuli. During walking, the right leg was found to be markedly spastic with moderate talipes equinovarus. No sensory alterations were found.
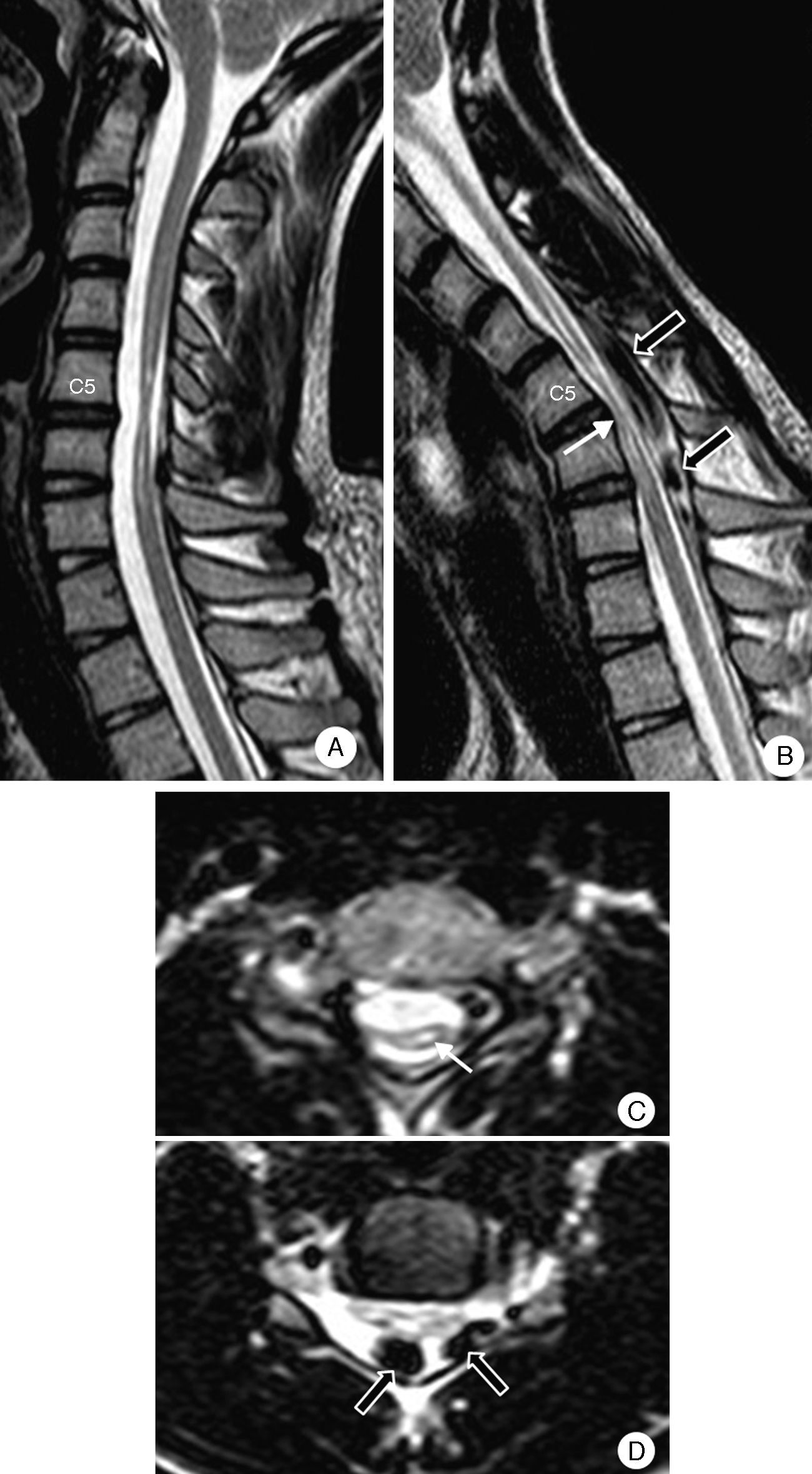
Complementary testsResults from a complete blood count and a biochemical analysis were normal. CSF was clear and contained no cells; glucose level was 60mg/dL, and protein level was 60mg/dL. Results from the Borrelia test and serology studies for Treponema pallidum, herpes virus, HIV, and human T-cell lymphotropic virus type 2 were negative. An EMG revealed active denervation and neurogenic deficits in the C7 and C8 territories. Results from the ENG and MRI studies were normal. A neutral position cervical MRI (Fig. 6) displayed segmental atrophy of the spinal cord at C5-C6 disc level and hyperintensities compatible with an intramedullary cavity extending from C5 to C7. In a flexed position, the scan showed spinal cord angulation at C5-C6, dura mater detachment from the posterior lamina, and large signal voids in the epidural space compatible with large dilated veins.
 Neutral position: severe atrophy at C5-C6 with spinal cord hyperintensity. (B) On flexion: the spinal cord presses against the edges of the vertebrae in the segment displaying the most atrophy (white arrow). Migration of the dura mater away from the posterior lamina and signal voids caused by venous engorgement (black arrows). (C) Neutral position (axial T2): severe asymmetrical predominantly right-sided atrophy, and hyperintensity compatible with spinal cord compression (arrow). (D) On flexion (axial T2): spinal cord pressing against a vertebra and large dilated epidural veins (black arrows).")
Patient 4. Cervical MRI scan. (A) Neutral position: severe atrophy at C5-C6 with spinal cord hyperintensity. (B) On flexion: the spinal cord presses against the edges of the vertebrae in the segment displaying the most atrophy (white arrow). Migration of the dura mater away from the posterior lamina and signal voids caused by venous engorgement (black arrows). (C) Neutral position (axial T2): severe asymmetrical predominantly right-sided atrophy, and hyperintensity compatible with spinal cord compression (arrow). (D) On flexion (axial T2): spinal cord pressing against a vertebra and large dilated epidural veins (black arrows).
Surgical treatment was suggested in view of the patient's age, rapid disease progression in just one year as reported by the family, and symptom severity (pyramidal signs and marked spinal cord atrophy). In February 2012, the patient underwent C5-C6 anterior discectomy and spinal fusion with graft, plate, and screws. The patient's condition has not worsened since his surgery, but rather remains stable after several months of follow-up.
DiscussionThe 4 cases presented here (all young males) displayed typical symptoms of Hirayama disease: asymmetrical weakness and atrophy of the hands and forearms with preserved brachioradialis, and insidious presentation in the second or third decades of life. The disease progressed slowly before stabilising in 3 of the patients.
Dynamic morphological alterations in cervical MR images are essential for a diagnosis of Hirayama disease. The MRI scan should be performed with the patient's neck at the optimal flexion angle. According to a systematic review, a neck flexion of 35° provides the greatest diagnostic sensitivity.10 Dura mater shifting on neck flexion is not enough to support an imaging-based diagnosis of Hirayama disease; this finding can also be seen in 46% of young asymptomatic controls.11 Diagnosis of the disease is based on the presence of stenosis with spinal cord compression and signal alterations in the epidural space (particularly a signal void indicative of dilated veins).
Although findings of venous congestion with neck flexion are essential for diagnosis, the role of this congestion in the pathogenesis of Hirayama disease is uncertain. Some authors have suggested that venous congestion may indicate reduced venous blood flow and result in spinal cord ischaemia due to stasis. However, although angiography studies12 have confirmed dilation of the epidural venous plexus, they failed to prove delays in either arterial or venous blood flow. Spinal cord lesions are therefore attributed to the mechanical factor in flexion myelopathy that leads to haemodynamic changes in spinal cord microcirculation.
The key debate for patients with Hirayama disease is how to proceed with treatment. The natural course of the disease seems to recommend conservative treatment, since disease progression stops after 2-3 years in many patients, and before 5 years in 75%. This seems to be true of our first 3 patients, especially patients 2 and 3: here, the anomalies in the epidural space that were seen at disease onset could no longer be seen in control MRI scans.
Treatment choice is more difficult in patients with particularly severe forms of the disease. During the first year after onset, Patient 4 presented severe spinal cord atrophy, amyotrophy extending to unusual segments (T1), highly active denervation with fasciculations, and clinical signs of pyramidal deficit suggestive of severe myelopathy. In similar cases, other doctors have also opted for surgical treatment.6 Since it remains unclear which surgical technique is best, we used both anterior spinal fusion9 and posterior duraplasty without spinal fusion.9,13 Articles report that surgery results in stabilisation of symptoms, except for one describing neurological improvements in only 3 out of 5 patients who underwent surgery.9
Given the rareness of Hirayama disease, its natural course, and the low numbers of patients undergoing surgery, it is very difficult to support surgical treatment. We feel that surgery must be considered only for severe cases presenting rapid disease progression.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Agundez M, Rouco I, Barcena J, Mateos B, Barredo J, Zarranz JJ. Enfermedad de Hirayama: ¿operar o no operar? Neurología. 2015;30:502–509.
