




Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) is an inherited adult-onset disorder characterised by migraine with or without aura, recurrent episodes of cerebral ischaemia without the typical cardiovascular risk factors, subcortical dementia, and neuropsychiatric disorders.1,2 Although this disease is underdiagnosed, it is considered the most frequent hereditary cause of ischaemic stroke.3
Doctors should suspect CADASIL in patients younger than 50 presenting typical symptoms, with a family history of similar entities, and whose brain MRI shows typical findings.3–5
We would like to present a new clinical case characterised by atypical presentation and absence of family history.
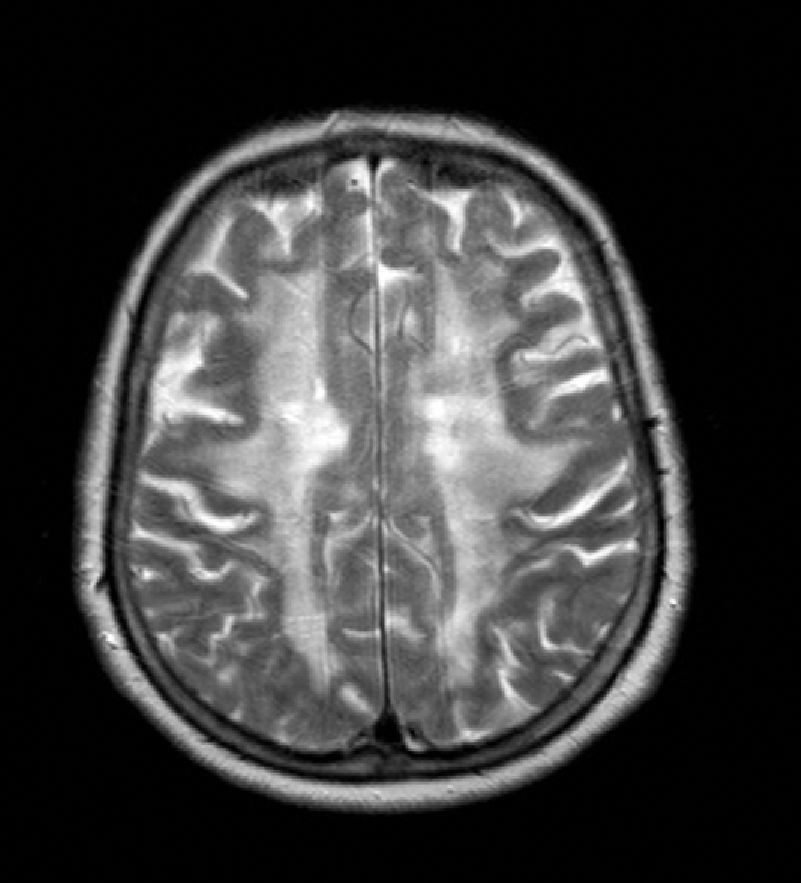
The patient was a 65-year-old woman with a history of transient and recurrent diplopia evolving over 30 years. In 1986, she was examined in another hospital, where lumbar puncture yielded normal biochemical and cytological findings and cranial CT revealed anomalies in the white matter. In the preceding 8 months, she had been experiencing symptoms of progressive apathy and amnesia. One month before the visit, she was referred to the neurology department due to a new episode of transient diplopia whose clinical characteristics were similar to those of prior episodes. Neurological examination yielded no relevant findings at that time and could not provide more information about the episodes, except that they always had the same presentation. Forty-eight hours before our consultation, she presented language difficulties, mental confusion, retropulsion when walking, and loss of sphincter control. Her personal history included dyslipidaemia, right-sided mixed hypoacusia, and left-sided neurosensory hypoacusia that required use of a hearing aid. The patient's parents had no history of stroke, migraine, or cognitive impairment. Examination showed the patient to be afebrile with a blood pressure of 130/70mm Hg. Orientation was limited to recognising people, and speech was halting and reduced to stereotyped word use. Comprehension was limited to simple commands and object naming; reading and writing abilities were impaired. Gait was characterised by retropulsion and wide-based stance accompanied by inability to walk unaided. Cranial nerves were spared and the confrontation visual field test was normal. Diplopia was not detected during the hospital stay. Physical examination revealed no other abnormalities. Blood test including a haemogram and basic biochemical assessment yielded normal results. Serum levels of calcium, thyroid hormones, vitamin B12, and folic acid were normal. Serology tests for HIV and syphilis were negative, as were results for antiphospholipid and antinuclear antibodies, and a thrombophilia study. Cranial CT displayed multiple ischaemic small-vessel lesions. CSF study showed normal results (1 cell, spinal fluid glucose concentration 70mg/dL, protein concentration 45mg/dL). Cranial MRI revealed extensive damage to bilateral supratentorial white matter, basal ganglia, and the bulbopontine region with presence of multiple ischaemic infarcts in basal ganglia (Fig. 1). MR angiography ruled out vascular malformations. The electroencephalogram, electromyogram, and visual evoked potentials were all normal. The ophthalmoscopy and carotid Doppler ultrasonography revealed no alterations.
.")
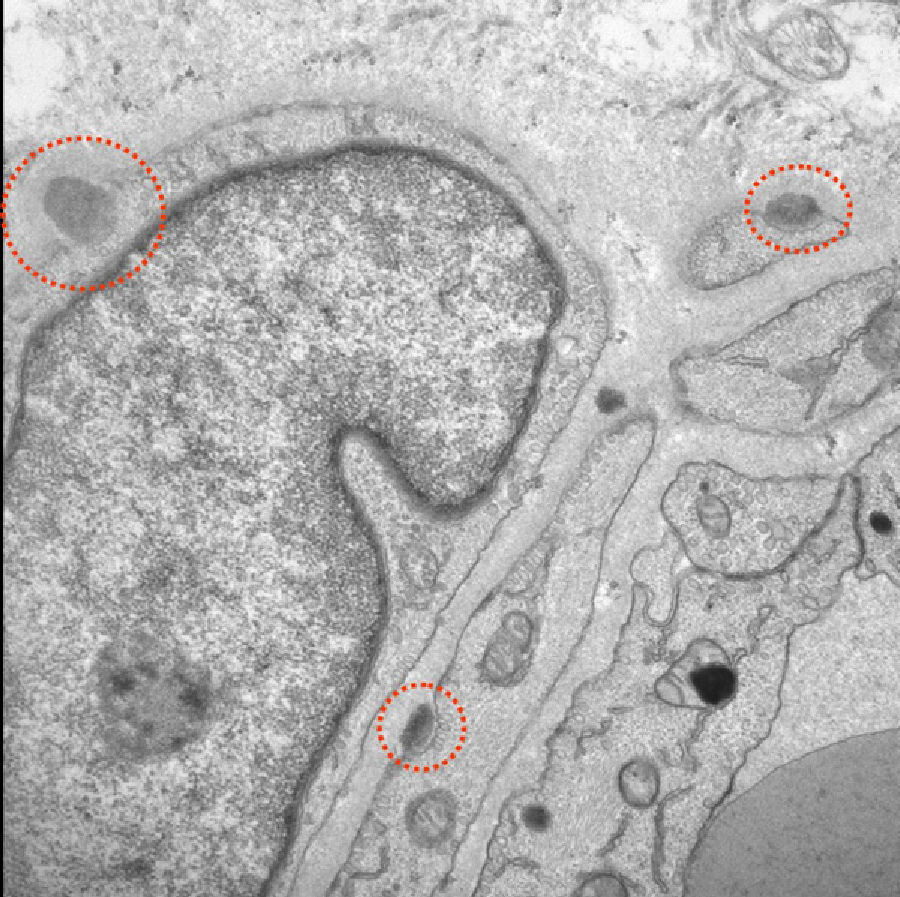
Given the absence of alternative diagnoses that could justify these findings, doctors performed a skin biopsy and a genetic study to rule out CADASIL, even though the patient did not meet classic criteria for this disease. Electron microscopy of the skin biopsy revealed granular electron-dense deposits. Typical of CADASIL, these deposits were located between smooth muscle cells, in cell indentations, or within the basement membrane (Fig. 2). The genetic study revealed a heterozygous sequence change (C>T) at position c.397 on the Notch3 gene which led to a p.Arg133Cys (A133c) amino acid variant. The patient was treated with acetylsalicylic acid and galantamine.

Doctors interviewed the patient's 3 asymptomatic children (2 daughters aged 34 and 41 years, and a 44-year-old son). None of her children had a history of migraines or other clinical manifestations compatible with their mother's diagnosis. MRI scans from the 2 daughters showed extensive damage to periventricular white matter and multiple subcortical infarcts; MRI in the son revealed only changes compatible with leukoaraiosis. Genetic tests and functional studies to confirm or rule out CADASIL are still pending. Genetic counselling was offered to the patient and cardiovascular risk factors were closely monitored.
CADASIL is a disease with various clinical manifestations and forms of presentation; this situation contributes to diagnostic difficulties and delays.6,7 The literature includes descriptions of visual disorders, especially amaurosis fugax, as initial manifestations of the disease.8,9 Likewise, researchers have published cases of early visual disorders that were detected by electrophysiological studies in asymptomatic patients with Cys146Tyr mutations.10 Diplopia is rare as a form of presentation of CADASIL. It has been mentioned as a manifestation of the brainstem impairment caused by the disease.4 However, the literature includes few clinical cases describing this symptom. Gurumukhani et al. published the case of a woman aged 52 who presented permanent diplopia in late stages of the disease. This was related to extensive white-matter damage.11 Blanco-Menendez et al. mentioned diplopia as the form of presentation of a transient ischaemic attack in the course of CADASIL.12 Lastly, Marrero-Falcon et al. described the case of a 55-year-old woman who, 10 years before, had begun experiencing recurrent episodes of vertigo and diplopia that were interpreted as transient ischaemia of the vertebrobasilar territory.13 In our case, diplopia appeared as the only symptom in similar recurrent and self-limiting episodes which she experienced over the years; between episodes, the patient's vision disorders resolved completely. This is unlikely to reflect transient ischaemic events in the posterior region of the brain, since such events are usually accompanied by other neurological deficits.14,15 Diplopia manifesting as a migraine aura is rare, but it has been described in the literature.16 This seems to be a case of aura without headache, since the patient has no history of headaches. According to the International Headache Society (IHS) classification, this is a subtype of migraine with aura.17 In spite of the fact that aura without headache has been described as a manifestation of CADASIL,18 no publications mention diplopia as an expression of that migraine subtype in this disease.
Regarding absence of family history of CADASIL, we should highlight that even if a positive family history supports the diagnosis of the disease, the absence of this background does not rule out this condition. De novo mutations may occur, and in such cases, there would be no detectable genetic alterations in the patient's ancestors. However, the mutation would be transmittable to the patient's descendants.19,20 Another relevant finding in our case was that diagnostic criteria (Table 1) showed low sensitivity, since they were unable to establish even a possible diagnosis for the patient. The decision to request tests to confirm CADASIL was based on typical MRI findings in the absence of an alternative aetiological diagnosis. We should also discuss the use of galantamine treatment in this case. Although no treatments have been shown to modify the course of the disease, some authors suggest that there may be a neuronal cholinergic deficit and that cholinomimetic drugs may be useful for the underlying cognitive decline. However, no randomised and controlled trials have explored this topic.21–23
Diagnostic criteria for CADASIL.
| Probable CADASIL |
| 1. Age at onset <50 years |
| 2. At least 2 of the following clinical findings: |
| • Stroke-like episodes with permanent neurological signs |
| • Migraine |
| • Major affective disorders |
| • Subcortical dementia |
| 3. Absence of cardiovascular risk factors aetiologically related to the deficit |
| 4. Evidence of an autosomal dominant inheritance pattern |
| 5. MRI showing white matter damage without cortical infarcts |
| Confirmed CADASIL |
| Criteria for probable CADASIL+discovery of Notch3 mutation and/or pathological findings showing small-vessel arteriopathy with granular osmiophilic material (GOM) deposits. |
| Possible CADASIL |
| 1. Late-onset (>45 years) |
| • Stroke-like episodes without permanent signs |
| • Minor affective disorder |
| • Global dementia |
| 3. Minor VRF: mild HTN, mild hyperlipidaemia, smoking, oral contraceptives |
| 4. Unknown or incomplete family history |
| 5. MRI with atypical changes in white matter |
| Exclusion criteria |
| 1. Age at onset >70 years |
| 2. Severe or complicated HTN with cardiac or systemic disease |
| 3. Absence of cases in a well-documented family tree |
| 4. Normal MRI at >35 years of age |
Lastly, the diagnosis may be confirmed in a patient's asymptomatic family members by means of a genetic study. This approach is frequently necessary in order to maximise preventive measures when cardiovascular risk factors are present and also to offer proper genetic counselling, given that the inheritance pattern is autosomal dominant. Although genetic study is the procedure of choice in asymptomatic patients, neuroimaging tests (especially MRI) play an important role since all carriers will develop the disease before the age of 60 and present typical imaging changes before the age of 40. If the brain MRI remains normal beyond this age, the subject is highly unlikely to develop the disease.5
In summary, CADASIL is a rare disease with a wide spectrum of clinical manifestations that have probably not yet been fully described. CADASIL should be considered in patients with recurrent episodes of diplopia and MR images that are typical of the disease. In addition, absence of family history is no reason to rule out this entity and not order tests that could confirm the diagnosis.
Please cite this article as: Rodriguez-Pecci MS, de la Fuente-Aguado J, Pato-Pato A, San Millan-Tejado B. Diplopía aislada como forma de presentación de CADASIL: a propósito de un caso. Neurología. 2014;29:56–59.
