



Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease (OMIM: 247100), is a rare recessive autosomal disorder. The disease follows a slow, benign course. To date, some 2501 to 300 cases2,3 have been reported. LP is characterised by intracellular deposition of periodic acid-Schiff–positive (PAS-positive) hyaline material in the skin, mucous membranes, and internal organs.4–6
This type of genodermatosis results from loss-of-function mutations in the gene coding for extracellular matrix protein 1 (ECM1) on chromosome 1q21.1,7–9ECM1 contains 10 exons with 3 isoforms (ECM1a, the most frequent; ECM1b; and ECM1c), whose functions remain to be determined. EMC1 is expressed in the dermis, keratinocytes, endothelial cells, and developing bones. It is linked to keratinocyte differentiation, basement membrane regulation, collagen composition, and growth-factor binding (skin homeostasis).1,9
Clinical manifestations of LP are secondary to protein abnormalities and vary greatly among individuals.5 The disorder affects multiple systems, especially the skin and mucosa of the upper aerodigestive tract.7 Nearly pathognomonic for LP, the disease typically presents in childhood with a weak cry and hoarse voice due to laryngeal infiltration.10 At the age of 3, infiltration and diffuse thickening of the skin occurs, resulting in papules and chickenpox-like scars. Around 50% to 60% of the cases present moniliform blepharosis resembling a string of pearls; this finding is almost pathognomonic.11 In 50% to 70% of the cases, cranial CT and MRI scans display bilateral, symmetrical calcifications in the medial region of the temporal lobes, including the hippocampus (unci) and the amygdalae12; other findings include epileptic activity, memory alterations, social/behavioural anomalies, paranoid symptoms, and mental retardation.1,8,12,13
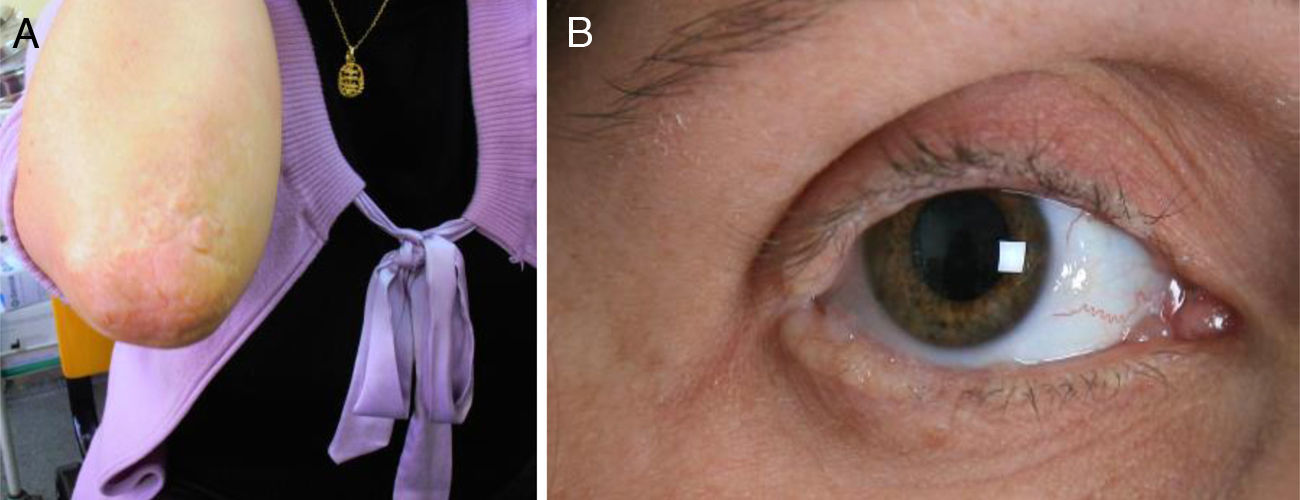
We present the case of a 35-year-old woman whose parents were not consanguineous. At the age of 9 months she displayed a hoarse, dysphonic voice and has visited our department since 1997 for numerous reasons: headache, memory loss, a feeling that she was experiencing things that were not real, difficulty recognising places, dizziness, instability, anxiety, and depression. She had a history of hypothyroidism. The physical examination revealed a dysphonic, low-pitched voice. The skin covering her joints displayed numerous yellowish hyperkeratotic verrucous papules in a paving-stone pattern; the lingual, labial, and jugal mucosae were also affected, and she displayed moniliform blepharosis (Fig. 1). The neurological examination was normal. The neuropsychological assessment revealed a slight decrease in information processing speed and mild alterations in episodic memory and recall processes.
 Hyperkeratotic plaques on the skin covering the elbows. (B) Moniliform blepharosis.")
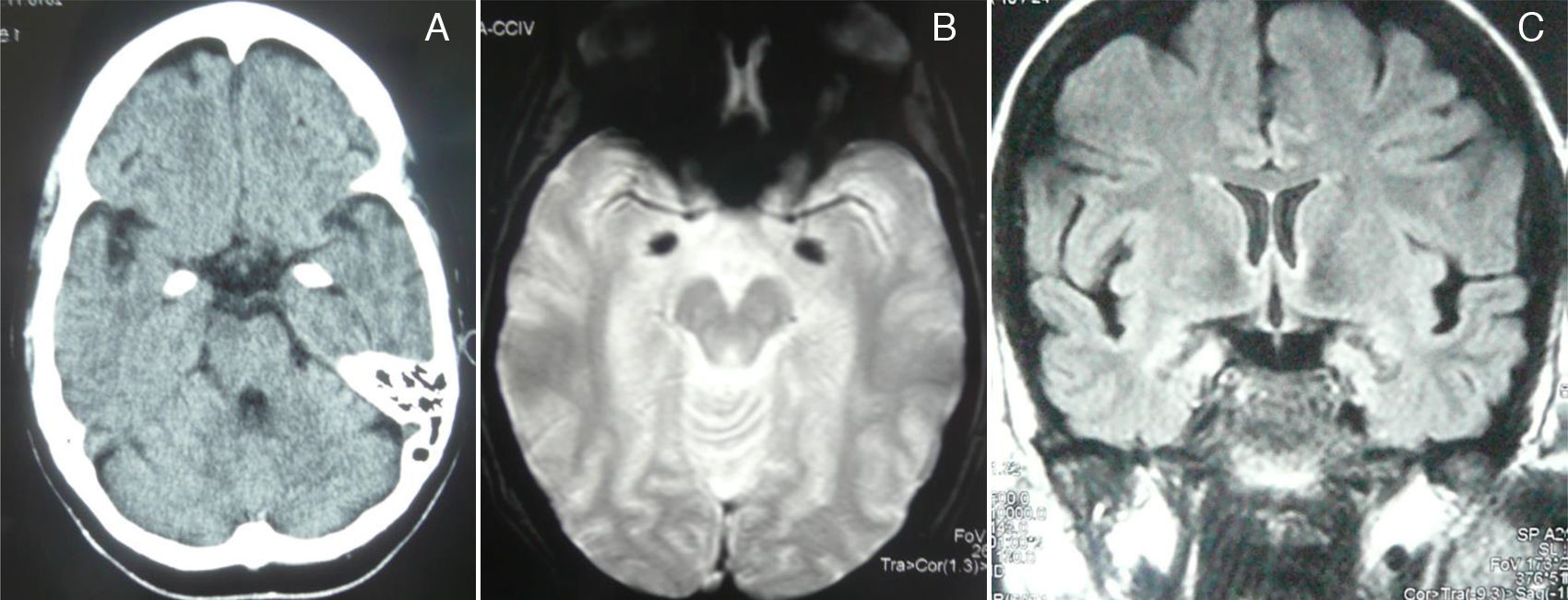
A complete analysis including hormone, antibody, immunity, and serology tests yielded no significant results. Cranial CT and MRI scans (T2*-weighted gradient-echo sequences) displayed bilateral, symmetrical hyperdense/hyperintense lesions in the unci and amigdalae, which were compatible with calcifications (Fig. 2). A sleep-deprived EEG revealed no abnormalities. Cognitive evoked potentials (P300) pointed to delayed reaction time in the Posner task and normal P3 latency in the oddball task.
, and coronal FLAIR MRI (hyperintensity) in the amigdalae and unci. Findings were compatible with calcifications.")
A skin biopsy revealed dermal and epidermal changes with irregular acanthosis, hyperkeratosis, and deposition of homogeneous eosinophilic PAS-positive diastase-resistant hyaline material around the blood vessels of the dermis and adjacent structures; these glycoprotein alterations were consistent with LP.
Sequencing the protein-coding region of the ECM1 gene revealed a nonsense mutation at exon 7 of ECM1, c.1076G>A, which resulted in a premature stop codon, p.Trp359*, affecting isoform ECM1a.
Further studies are necessary to establish a more specific connection between genotype and phenotype; in our case, we hypothesised that the new mutation may have resulted in a more extensive phenotype with skin, mucosa, and brain involvement. Likewise, mutations outside exon 7 (such as those affecting the ECM1b isoform, which lacks exon 7) have been associated with a more severe mucocutaneous phenotype but no neurological involvement.14
To the best of our knowledge, this is the first case of genetically confirmed LP with brain calcifications to be published in the Spanish-language literature.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Abril-Jaramillo J, Mondéjar R, Lucas M, García-Bravo B, Ríos-Martin JJ, García-Moreno JM. Lipoidoproteinosis o enfermedad de Urbach-Wiethe: a propósito de un nuevo caso con afectación cerebral. Neurología. 2017;32:125–127.
The genetic part of this study has been published previously (J Clin Neurol. 2014;10:64–8).

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas