



La multineuritis craneal se define como afectación de varios pares craneales e implica un diagnóstico diferencial complejo entre múltiples etiologías1,2.
Una de las etiologías de la neuropatía craneal es la neurolinfomatosis, que se define como la infiltración por células linfoides malignas de los nervios ya sean periféricos, espinales, pares craneales o plexos, con afectación en múltiples territorios3,4. Se relaciona en un 90% con linfomas no-Hodgkin y un 10% con leucemia mal diferenciada5. Puede presentarse como debut del proceso oncológico (primaria)6,7. La neurolinfomatosis primaria es una entidad poco frecuente, existe escasa información sobre su forma de presentación, curso, diagnóstico y tratamiento, de ahí lo interesante del caso.
Presentamos el caso de un varón de 36 años sin antecedentes. Inició de forma progresiva en tres meses con un cuadro de parálisis del VI nervio craneal izquierdo, parálisis facial periférica izquierda con mejoría posterior y parálisis facial periférica derecha. En el ingreso presentó a la exploración neurológica afectación de nervios craneales (ambos VI nervios craneales, VII nervio craneal derecho), sin otros hallazgos. El estudio inicial no mostró alteraciones. Este consistió en pruebas de neuroimagen (tomografía computarizada [TC] cerebral, resonancia magnética cerebral con contraste y de base de cráneo), analítica completa (bioquímica, estudio microbiológico, marcadores tumorales, estudio de autoinmunidad, enzima convertidora de angiotensina [ECA], vitamina B12, ácido fólico, hormonas tiroideas, proteinograma, electroforesis, marcadores tumorales, serologías, test antigénico rápido y reacción en cadena de la polimerasa para SARS-CoV-2 y estudio de autoinmunidad) así como ecografía testicular y tiroidea.
Se realizó punción lumbar para análisis de líquido cefalorraquídeo (LCR), objetivándose pleocitosis linfocitaria (68 células /mm3, Glucosa 51 mg/dL, proteínas totales 38,00 mg/dL), siendo el resto de los estudios (citobioquímica, ECA, bandas oligoclonales, microbiológico, tinción de Gram, cultivos y serologías) negativos.
La mayoría de los linfocitos del LCR mostraban una morfología atípica al examinarlos al microscopio óptico y en el análisis por citometría de flujo (CF) se observaba una población anormal de linfocitos que expresaban un inmunofenotipo compatible con leucemia/linfoma linfoblástico T.
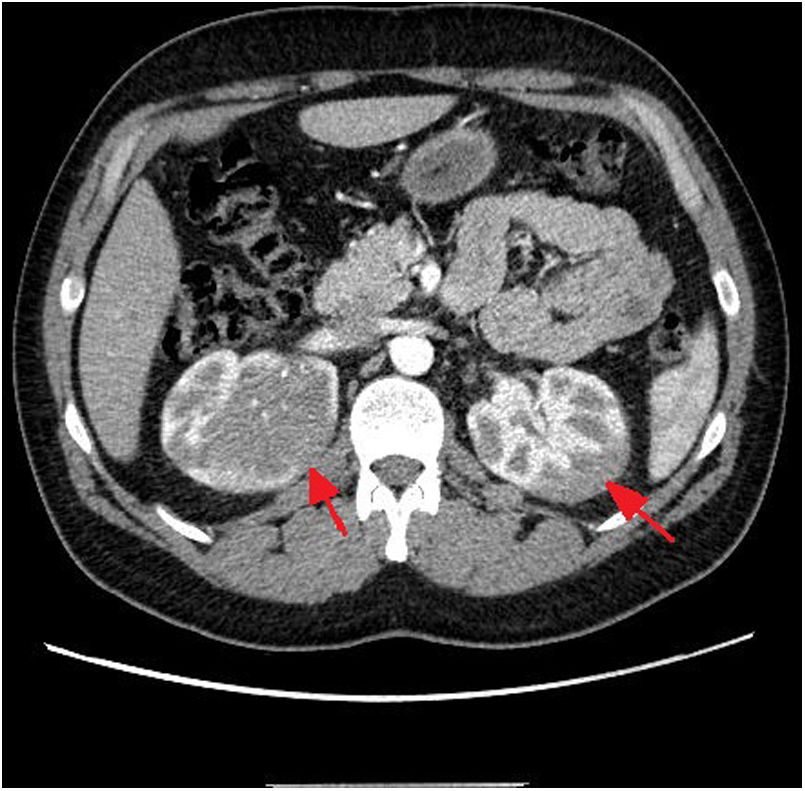
Dados los hallazgos, se amplió estudio con TC toraco-abdomino-pélvico donde se objetivó masa renal bilateral de carácter infiltrativo (fig. 1). Se realizó biopsia con aguja gruesa de masa renal derecha y el estudio anatomopatológico confirmó infiltración del parénquima renal por una población linfoide atípica con alto índice proliferativo compatible con linfoma linfoblástico T. La biopsia de médula ósea mostró leve infiltración (1,6%). Se inició tratamiento con dexametasona 6 mg cada seis horas.

Tras diagnóstico definitivo de linfoma linfoblástico T se inició triple terapia intratecal (TIT) con metotrexato, citarabina e hidrocortisona, junto a quimioterapia sistémica intensiva siguiendo protocolo de linfoma agudo linfoblástico 20118.
El paciente presentó buena respuesta al tratamiento, con mejoría progresiva de la clínica neurológica hasta quedar asintomático. Tras un ciclo de TIT el LCR fue acelular y no se detectó enfermedad por CF. La TC de reevaluación mostró resolución casi completa de las masas renales. El paciente pasó a cargo de Hematología con resolución de la clínica neurológica.
La multineuritis craneal es una entidad que requiere buena integración clínica y estudio completo, realizando un amplio diagnóstico diferencial entre múltiples etiologías (tumorales, infecciosas, vasculares, autoinmunes)9. Esta clínica puede presentarse por afectación en cualquier lugar anatómico desde una alteración en troncoencéfalo hasta disfunción de nervio a nivel periférico. Una vez identificada la etiología que produce el cuadro, el manejo terapéutico de la multineuritis craneal consiste en el tratamiento específico de la enfermedad de base9.
Una de las etiologías poco frecuentes causante de esta clínica es la neurolinfomatosis. La neurolinfomatsis puede preceder a la enfermedad sistémica hasta en un cuarto de los pacientes6. Por lo poco frecuente de esta presentación en enfermedades hematológicas malignas, el diagnóstico en ocasiones se retrasa y la incidencia de esta patología por el momento se desconoce5. La heterogeneidad clínica y la inespecificidad en las pruebas de imagen cerebral, sumado a la escasa rentabilidad del análisis del LCR (positividad de células linfomatosas sólo en un 20-40% de los casos) pueden condicionar un infradiagnóstico y retraso en el tratamiento del proceso hematológico de base. Un tratamiento temprano de la patología de base conlleva mejor pronóstico10.
Por el momento no existe evidencia sobre el tratamiento óptimo, según la literatura consiste en quimioterapia intensiva adaptada al tipo de linfoma primario o la recidiva del mismo5,6. En el caso de neurolinfomatosis en los linfomas linfoblásticos T8,11 las estrategias terapéuticas que se emplean son las mismas que en las leucemias agudas linfoblásticas12,13. La elección del tratamiento debe establecerse por consenso de hematólogos experimentados.
A nivel pronóstico la mayoría responde bien a la quimioterapia inicial obteniéndose buena funcionalidad tras la misma, a pesar de ello a largo plazo el pronóstico es pobre, con una supervivencia media de 10 meses, pudiendo ser algo mayor en el caso de las linfomatosis primarias4.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

