



Methylmalonic acidaemia with homocystinuria is an infrequent inborn error of vitamin B12 (cobalamin) metabolism. It is caused by defects in the synthesis of the coenzymes adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl), leading to decreased activity of the corresponding enzymes methylmalonyl-CoA mutase (MUT;609058) and 5-methyltetrahydrofolate-homocysteine methyltransferase, also known as methionine synthase (MTR;156570). Four complementation classes of cobalamin defects (cblC, cblD, cblF, and cblJ, caused by mutations in the corresponding genes) are responsible for methylmalonic acidaemia with homocystinuria. Methylmalonic acidaemia with homocystinuria type cblD is caused by mutations in the MMADHC gene (2q23.2), following an autosomal recessive inheritance pattern. We present 2 cases.
Patient 1 is an 18-year-old woman of Roma origin. The patient had no relevant medical history and did not follow a specific diet, and was admitted due to fever of 38.5°C, sudden-onset confusional symptoms, disorientation in time and space, incoherent but non-dysarthric speech, and psychomotor delay. She displayed a good level of consciousness; she was alert, disoriented, anxious, and irritable but not aggressive. The patient presented no headache or meningeal signs. A blood test, biochemical test, total protein test, immunoglobulins, autoimmunity study, serological test, and tests for thyroid hormone levels, ferritin, vitamin B12, and folic acid all yielded normal results. Results of a head CT scan and lumbar puncture were normal (negative microbiological study).
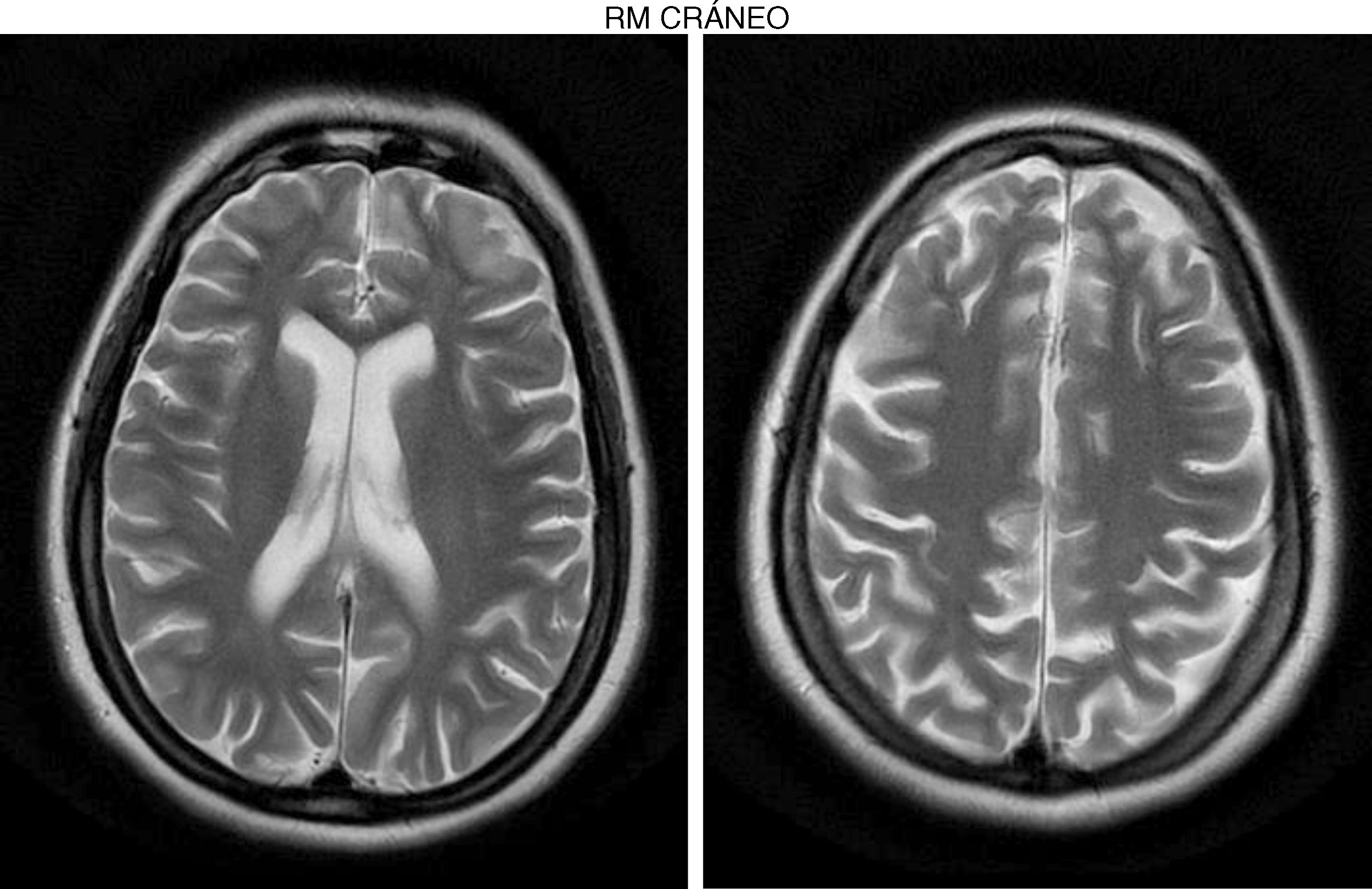
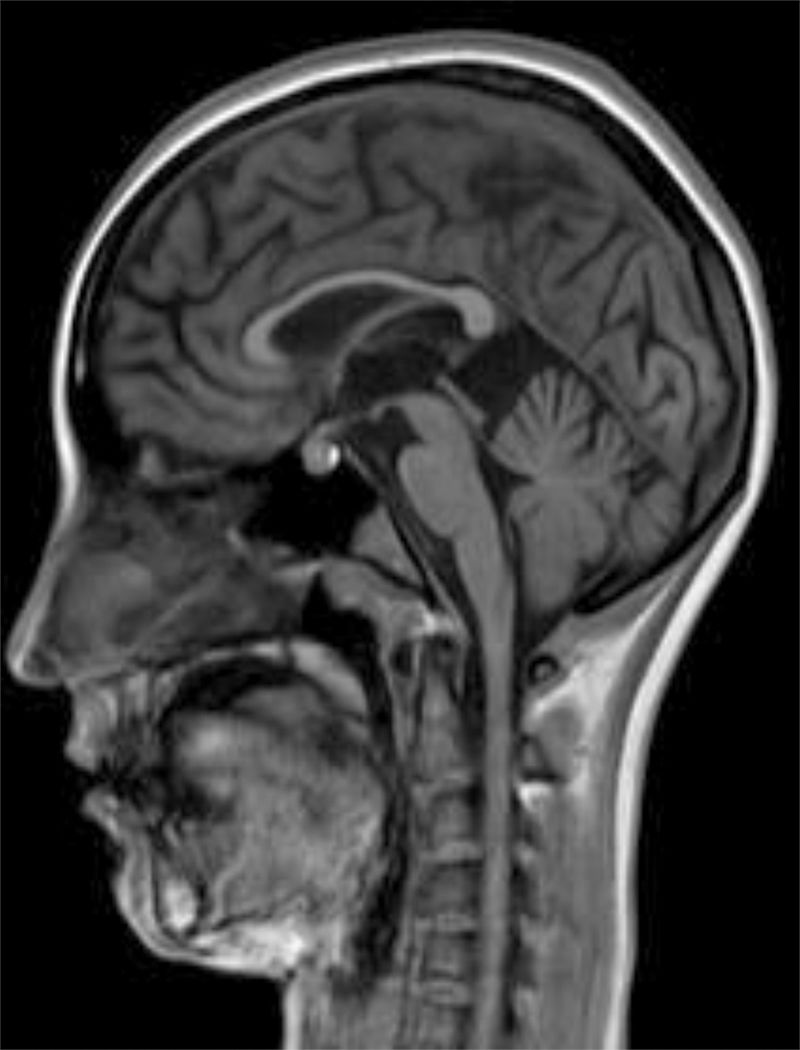
The internal medicine department prescribed empirical treatment with aciclovir, which achieved favourable progression until all symptoms disappeared. She did not attend the follow-up appointments scheduled at discharge, and consulted 10 months later due to gradual, progressive gait disorder (weakness in the lower limbs), frequent falls, and loss of strength in the upper limbs. During the examination, she was alert and presented mild dysarthria, pronounced bradypsychia, and difficulty maintaining attention. The patient displayed frontal release signs. Pupils were isocoric and reactive. Examination revealed muscle strength of 3/5 in the psoas, 3/5 in the quadriceps, 4/5 in the gluteus medius, 3/5 in the gluteus maximus, 2/5 in the tibialis anterior, 1/5 in the extensor digitorum muscle, 3/5 in the hamstring, and 1/5 in the ankle dorsiflexor. We identified patellar tendon hyporeflexia (2/4) and bilateral ankle clonus. In the upper limbs, reflexes were 3/4 and strength 4/5. Deep sensibility was affected in distal areas; hypoaesthesia was also observed. We also observed ataxic gait, grade I spasticity, and bilateral Babinski sign. A brain MRI scan (Fig. 1) showed cortical retraction, slight dilation of the subarachnoid spaces of the convexity, and discrete enlargement of the supratentorial ventricular system with thinning of the corpus callosum. An EEG revealed slow background activity with no epileptiform abnormalities. A motor nerve conduction study showed a discrete sensorimotor axonal polyneuropathy predominantly affecting the lower limbs. The differential diagnosis included the study of hereditary spastic paraparesis, which yielded negative results. Furthermore, autoimmune and paraneoplastic disease screening also detected no abnormalities. We then decided to study late-onset inborn errors of metabolism and requested blood homocysteine and blood and 24-h urine amino acid and organic acid levels; results showed increased methylmalonic acid (258nmol/mol of creatinine, normal: <0.56) and methylcitric acid and slightly elevated ketones (3-hydroxybutyric acid) in the urine, as well as increased blood homocysteine (339.2μmol/L, normal: <10). During the study, the patient presented popliteal vein thrombosis with massive bilateral pulmonary thromboembolism, which led to the patient's death.

Patient 2 is an 15-year-old woman of Roma origin, sister of patient 1 (3 years younger).
Development was normal. Several months before her sister's death, she presented insidious symptoms of difficulty walking, loss of appetite, and inappropriate laughter. Examination revealed bradypsychia and difficulty maintaining attention, ataxic gait with generalised spasticity (grade I), impaired hip adduction, mild genu recurvatum, muscle strength of 4/5 in the psoas, 3/5 in the quadriceps, 1/5 on dorsiflexion, sustained soleus clonus when seated (nonsustained when standing), and mild ataxia of the left arm. We observed symmetrically increased deep tendon reflexes in the lower limbs and bilateral Babinski sign. Muscle strength was 4/5 proximal and 5/5 distal in the upper limbs. Eye fundus and MRI findings were similar to those observed in her sister (Fig. 2). An EEG revealed slow background activity. She presented 2 generalised tonic-clonic seizures with no apparent trigger, which required antiepileptic treatment.

The determination of amino acid and organic acid levels showed increased blood homocysteine (169.9μmol/L, normal: <10) and urine amthylmalonic acid (15.40nmol/mol of creatinine, normal: <0.56). The study of genes involved in methylmalonic acidaemia with homocystinuria detected a homozygous mutation (c.748C>T) in the MMADHC gene (found in the DNA of both sisters), which generates a stop codon that results in a truncated protein, provoking the disease.1–3 The patient is currently receiving treatment, and presents significant improvement and neurological stability.
ConclusionDefinitive diagnosis of these patients enables the early introduction of a specific treatment, as well as genetic counselling and, where necessary, prenatal diagnosis.
In patients presenting symptoms of paraparesis, axonal and demyelinating polyneuropathies, cognitive impairment of unknown cause, and deep venous thrombosis, we should analyse homocysteine and vitamin B12 metabolism.4,5
Informed consentThis article was submitted for publication with the written consent of the patients’ parents.
Authors’ contributionsDr E. Cancho García established clinical diagnosis and treatment. Dr E. Geán and Dr T. Oliver Tormo performed the genetic/metabolic study. Dr Torrents performed the metabolic study. Dr E. Esteban Durán was responsible for the radiology studies.
Please cite this article as: Cancho García E, Geán E, Oliver Tormo B, Torrents A, Esteban Durán E. Mutación del gen MMADHC de la cobalamina D con comienzo en el adulto: a propósito de 2 casos potencialmente tratables. Neurología. 2019;34:419–421.
