



Overpopulation and industrial growth result in an increase in air pollution, mainly due to suspended particulate matter and the formation of ozone. Repeated exposure to low doses of ozone, such as on a day with high air pollution levels, results in a state of chronic oxidative stress, causing the loss of dendritic spines, alterations in cerebral plasticity and in learning and memory mechanisms, and neuronal death and a loss of brain repair capacity. This has a direct impact on human health, increasing the incidence of chronic and degenerative diseases.
DevelopmentWe performed a search of the PubMed, Scopus, and Google Scholar databases for original articles and reviews published between 2000 and 2018 and addressing the main consequences of ozone exposure on synaptic plasticity, information processing in cognitive processes, and the alterations that may lead to the development of neurodegenerative diseases.
ConclusionsThis review describes one of the pathophysiological mechanisms of the effect of repeated exposure to low doses of ozone, which causes loss of synaptic plasticity by producing a state of chronic oxidative stress. This brain function is key to both information processing and the generation of structural changes in neuronal populations. We also address the effect of chronic ozone exposure on brain tissue and the close relationship between ozone pollution and the appearance and progression of neurodegenerative diseases.
La sobrepoblación y el crecimiento industrial causan un aumento en la contaminación del aire, principalmente de partículas suspendidas y en la formación de ozono. La exposición repetida a bajas dosis de ozono, como la de un día con alta contaminación del aire, genera un estado de estrés oxidativo crónico, el cual causa pérdida de espinas dendríticas, alteraciones en la plasticidad cerebral y en los mecanismos de aprendizaje y memoria, así como muerte neuronal y pérdida de la capacidad de reparación cerebral. Esto tiene un impacto directo en la salud humana, aumentando la incidencia de enfermedades crónico-degenerativas.
DesarrolloSe realizó una búsqueda de artículos originales y revisiones en Pubmed, Scopus y Google Scholar (2000-2018) sobre las principales consecuencias de la exposición a ozono, sobre la plasticidad sináptica, sobre el procesamiento de la información en los procesos cognitivos y sobre las alteraciones que pueden llevar al desarrollo de enfermedades neurodegenerativas.
ConclusionesEsta revisión describe uno de los mecanismos fisiopatológicos del efecto de la exposición repetida a bajas dosis de ozono, a través de producir un estado de estrés oxidativo crónico, lo cual causa pérdida de la plasticidad sináptica; esta función cerebral que es clave en tanto el procesamiento de información como en la generación de cambios estructurales en las poblaciones neuronales. También se aborda el efecto de la exposición crónica a ozono sobre el tejido cerebral, y la estrecha relación entre la contaminación por ozono y la aparición y evolución de las enfermedades neurodegenerativas.
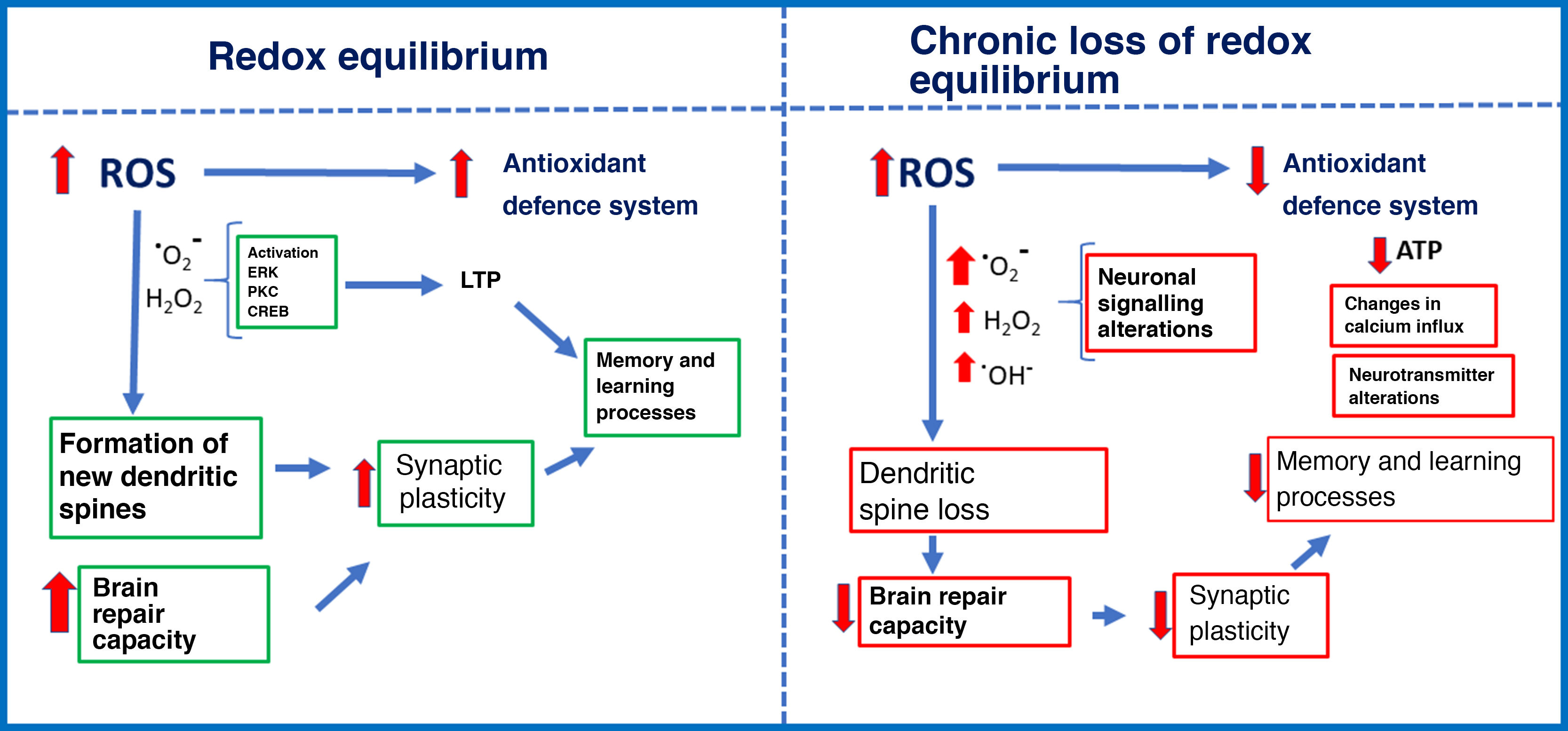
Repeated exposure to low doses of ozone, as occurs on days of high pollution, causes a state of chronic oxidative stress. The effects of solar radiation on gas pollutants (carbon monoxide and dioxide, nitric oxide, and volatile organic compounds) produces such molecules as ozone, which have a high oxidation potential. Chronic exposure to ozone causes oxidative stress, with negative consequences including elevated levels of reactive oxygen species (ROS) and decreased response of the antioxidant defence system. Oxidative stress may be defined as an imbalance between the rate of oxidant production and the rate of oxidant degradation by the antioxidant defence system.1 In redox equilibrium, oxidant signals act as intracellular and extracellular signalling molecules to regulate cell metabolism and the immune response. Aerobic organisms, mainly vertebrates, obtain energy through the oxidation of organic substrates (glucose) with molecular oxygen. Sometimes, however, oxygen is partially reduced, resulting in the generation of such pro-oxidant molecules as the superoxide ion and highly reactive intermediates.2 These intermediates are easily neutralised by the antioxidant defence system, maintaining the balance of oxidation and reduction. However, elevated ROS levels and antioxidant defence system dysfunction cause a state of chronic oxidative stress3 that alters cellular processes, resulting in organelle damage, metabolic alterations, and the onset of apoptosis (Fig. 1).1
 and chronic loss of redox equilibrium (right).")
ROS include free radicals and their metabolites, such as superoxide and hydroxyl radicals and hydrogen peroxide (H2O2). These ROS can oxidise DNA,4,5 proteins, and lipids.5,6
ROS are generated by both endogenous and exogenous mechanisms. Endogenous ROS are produced during mitochondrial aerobic metabolism in the endoplasmic reticulum and in peroxisomes.7 Furthermore, increased ROS production is accompanied by elevated levels of reactive nitrogen species. Exogenous ROS, in contrast, result from exposure to a number of environmental factors, including air pollution and oxidants resulting from combustion of hydrocarbons and tobacco (these interact with skin molecules and can even cross the skin barrier, reaching the bloodstream8,9), and solar UV radiation, which is absorbed by the skin and induces the formation of numerous pro-oxidants.10
Reactive oxygen speciesOxygen is essential for aerobic metabolism. However, it may also be toxic under certain circumstances. During mitochondrial metabolism, oxygen is used in a series of chemical reactions that produce energy. The process also produces free radicals, which are neutralised by such antioxidant defence mechanisms as superoxide dismutase (SOD) and glutathione through a series of oxidation-reduction reactions in which water is produced through the transfer of electrons.11,12
The effects of ROS depend on the concentration. At physiological concentrations during redox equilibrium, ROS signal metabolic responses in cells. Increased ROS levels stimulate the antioxidant defence system, promote cell growth, regulate the cell cycle, modulate the immune system, etc.13,14 However, low ROS concentrations due to chronic ozone exposure alter the antioxidant defence systems, leading to impaired intracellular signalling and cell metabolism, and apoptosis.7,15–17 We should stress that oxidant signals, both at physiological levels and at increased concentrations, induce an endogenous antioxidant response. However, repeated exposure to low concentrations renders the antioxidant defence system unable to fight chronic oxidative stress. This may explain the dual action of ozone: high-dose ozone can be administered for therapeutic purposes, but chronic low-dose exposure is associated with degenerative disease.
The antioxidant defence systemDuring the evolution of life, the appearance of pigments gave rise to photosynthetic organisms, which increased atmospheric oxygen gas concentrations, enabling the development of the mechanisms necessary to use the molecule through the oxidation of glucose, producing great quantities of energy in the form of adenosine triphosphate (ATP) and generating ROS. This provided cells with highly efficient energy production systems. The evolutionary advantage of aerobic organisms also increased levels of free radicals and ROS, and led to the development of antioxidant defence systems.
These systems are formed by low–molecular-weight substances able to spontaneously neutralise ROS and their derivatives, and can act in various ways: 1) decreasing the concentration of oxidants; 2) preventing the reaction through free radical scavenging; 3) binding to metal ions to prevent the formation of ROS; 4) converting peroxides into less reactive products; and 5) halting the propagation and increase of free radicals.12,18 Antioxidant defence systems help maintain redox equilibrium, acting as a molecular barrier to protect against oxidative stress.19
Proteins that bind to transition metals, for instance transferrin, ferritin, and ceruloplasmin, confer protection as they limit the metal’s ability to participate in oxidative reactions.20 Furthermore, some endogenous enzymes, including SOD, catalase, glutathione reductase, and glutathione peroxidase, prevent the formation of oxidants. These enzymes serve as the first-line antioxidant defence against primary oxidants. In addition to these, such other antioxidant enzymes as lipolytic and proteolytic enzymes and glutathione S-transferase enzymes can act as ROS scavengers. In physiological conditions, cells are able to defend against high concentrations of ROS by achieving a balance between ROS production and ROS neutralisation.21–23
Such dietary antioxidants as β-carotene (a vitamin A precursor), retinoic acid, and ascorbic acid (vitamin C) neutralise and scavenge O2− and maintain α-tocopherol (vitamin E) in the reduced, active state. α-Tocopherol arrests the chain reaction of lipid and glutathione peroxidation, whereas other thiols (e.g., dihydrolipoate) block H2O2 and contribute to maintaining tocopherol and ascorbic acid in the reduced form.12,24–26
Pollution and models of oxidative stress caused by reactive oxygen speciesCurrent research into oxidative stress follows 2 different experimental approaches: the production of either exogenous or endogenous ROS. In the first approach, the animal model is exposed to an oxidant environment, such as environmental pollution; for example, animals may be repeatedly exposed to low doses of ozone7,16,27 or administered such toxic substances as paraquat, which induce chronic oxidative stress by generating free radicals28,29 and depleting the levels of antioxidants. In the second approach, oxidative stress is induced with the overproduction of pro-oxidant molecules, as is the case with the 3-nitropropionic acid model; this excitotoxin inhibits mitochondrial respiration and reduces ATP levels, causing oxidative stress and inhibiting antioxidant production.30,31 Other studies have used transgenic models.32–34
Furthermore, several animal species have been used to study the effects of ROS. Some examples are such invertebrates as the fruit fly Drosophila melanogaster and the nematode Caenorhabditis elegans, in which point mutations induced in specific genes have been used to modify the amount of SOD and catalase molecules available in different tissues.35–37 Other models of oxidative stress have been developed in mammals including rats, mice, and monkeys. Animal models have enabled the study of oxidative stress both in vitro and in vivo.
These studies have shown that oxidative stress has multiple harmful effects, particularly neurotoxicity, which manifests as neuronal dysfunction and degeneration and cell death.
Cell cultures can also be used to measure changes in the oxidant status of neurons; this approach can be used to study the role of oxidative stress in disease pathogenesis.38–40 Another methodological approach studies the increased production of free radicals with molecular fluorescence tests that are sensitive to oxidation, such as dichlorodihydrofluorescein diacetate staining, enabling the detection of intracellular ROS production.41–43 This technique has been used in primary cultures of rat hippocampal neurons; these studies identified pro-oxidant molecules with fluorescence microscopy.44
Studies with animal models replicating the environmental pollution to which humans may be exposed have revealed the significant role of chronic oxidative stress in disease pathogenesis: a state of chronic oxidative stress alters cell signalling, leading to dysregulation of the inflammatory response and alterations in intracellular signalling pathways involved in regulating cell function. These alterations are involved in the complex mechanisms causing neurodegenerative diseases. Furthermore, chronic exposure to low doses of ozone is a useful non-invasive model for studying the role of oxidative stress in neurodegeneration; there is evidence that oxidative stress alone is capable of causing progressive neurodegeneration in the hippocampus of rats exposed to ozone.45 Ozone-induced oxidative stress results in an increase in ROS,7 which induce cell damage46 and lipid peroxidation in various brain regions.45,47 They also cause mitochondrial dysfunction,48 endoplasmic reticulum stress,16 increased microglial activation,45 increased IL-17A levels,17 and neuronal death, depending on exposure duration.45,49
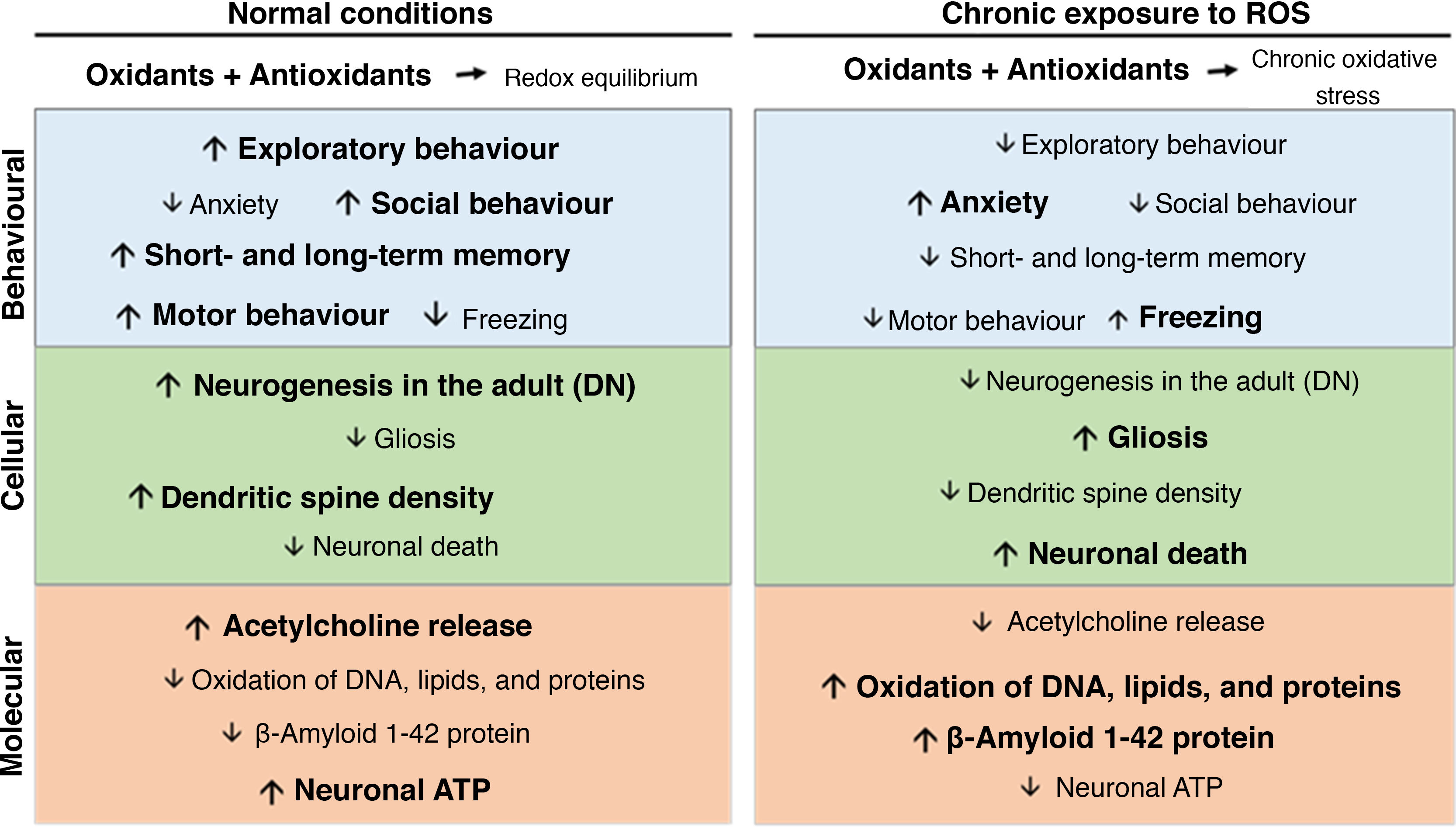
Oxidative stress, synaptic plasticity, and neurodegenerative diseaseChronic oxidative stress secondary to chronic ozone exposure negatively affects behaviour and cognitive function. Chronic ozone exposure inhibits exploratory behaviour and increases freezing behaviour in rodents,50,51 decreases the percentage of time spent on the open arms of the elevated plus maze and in social interaction, and reduces climbing behaviour.52 Chronic ozone exposure has also been found to affect both short-term and long-term memory in passive avoidance tasks (Fig. 2).15

At the cellular level, chronic ozone exposure inhibits neurogenesis in the dentate gyrus of adult animals, in addition to promoting gliosis and increasing the numbers of phagocytic microglia.45 Colín-Barenque et al.53 studied the effects of acute ozone exposure and found significant decreases in dendritic spine density in the rat olfactory bulb 24 hours after exposure; these effects were still visible 10 days after exposure. Other studies report decreased dendritic spine density in secondary and tertiary dendrites of the striatum and in pyramidal neurons of the prefrontal cortex.50 At the level of neurotransmission, chronic ozone exposure decreases the levels of acetylcholine, acetylcholinesterase, and choline acetyltransferase in the CA3 region of the rat hippocampus (Fig. 2).15
Some ROS play an essential role in signalling pathways involved in synaptic plasticity in several brain regions. In the hippocampus, superoxide anion is necessary to activate such protein kinases as the extracellular signal-regulated kinase and protein kinase C during long-term potentiation induction.54 However, presence of antioxidants and superoxide scavengers prevents the activation of the extracellular signal-regulated kinase in hippocampal slices.55 This leads to the blockage of several signalling pathways that trigger synaptic plasticity–related gene expression.
Other studies have shown that H2O2 is essential in the phosphorylation of extracellular signal-regulated kinase and cAMP response element–binding protein in response to NMDA receptor stimulation in the hippocampus.56,57 H2O2 is also involved in Ca2+ release from intracellular reservoirs, which enables the activation of signalling cascades that promote the expression of genes involved in information processing. This may explain why regenerative therapy with high doses of ozone or other pro-oxidant agents is beneficial for neuronal health, provided that the antioxidant defence system is active. However, low-dose ozone has also been observed to cause excessive oxidant production due to antioxidant defence system dysfunction; this is highly toxic for synaptic plasticity, and usually has a direct impact on cognition.
The fate of brain synapses depends on the redox equilibrium. In a pro-oxidant cell environment, ROS will cause dendritic spine pruning; in an antioxidant environment, in contrast, the effects of ROS are neutralised and synapses are preserved.58
Although ROS are known to act on some molecules actively involved in plasticity, very little is known about the effects of these pro-oxidant molecules on the changes in neuronal structure underlying synaptic plasticity. Some areas of the human brain, such as the basal ganglia and the substantia nigra, contain large amounts of iron and other transition metals that play an essential role in ROS production.59,60
The role of reactive oxygen species in Alzheimer diseaseStudies of primary cultures of rat cortical cells have shown that β-amyloid decreases the activity of complex I (NADH dehydrogenase) and complex IV (cytochrome c oxidase). Complex I is considered the main promoter of ROS in normal physiological conditions; alterations in complex I activity may result in increased production of ROS and induce total energy consumption due to disruption of oxidative phosphorylation. In Alzheimer disease (AD), mitochondrial alterations caused by β-amyloid overproduction induce ROS production, leading to cell damage and death.61,62 Neuronal ATP depletion results in neurotransmission dysfunction and impaired axonal transport. It also causes dysfunction in ATP-dependent ion channels, altering ion balance in the cytosol.61
In the rat hippocampus, oxidative stress causes progressive neurodegeneration, which leads to neuronal death, loss of neurogenesis, glial activation, increased β-amyloid 1-42 synthesis, and structural changes in β-amyloid 1-42.27,45
Furthermore, studies with animal models and humans have shown a prolonged increase in the pro-oxidant state throughout the course of AD; these conclusions are based on evidence that ROS cause changes in the redox equilibrium of the ageing brain, which may contribute to the pathogenesis of the disease.63–65 Weakening of the phospholipid membrane (one of the main consequences of lipid peroxidation) has been found to play a role in the pathogenesis of such neurodegenerative diseases as AD.66 β-Amyloid favours lipid peroxidation. Increased levels of lipid peroxidation products and reduced antioxidant activity are strongly correlated with the formation of senile plaques and neurofibrillary tangles in the brains of patients with AD. Furthermore, markers of oxidative stress have been detected in brain tissue and cerebrospinal fluid in these patients.67,68
In Parkinson’s disease (PD), oxidative stress is widely associated with increased dopamine and iron oxidation and reduced glutathione levels, causing a vicious circle that maintains the state of oxidative stress and destroys dopaminergic neurons.46,69 PD is characterised by α-synuclein accumulation and gradual loss of dopaminergic neurons in the substantia nigra.70,71 The disease is associated with increased levels of 8-hydroxydeoxyguanosine, which has been linked to increased mitochondrial DNA deletions in unaffected dopaminergic neurons of the substantia nigra, and in the serum.72,73 These patients display lower concentrations of polyunsaturated fatty acids and increased levels of lipid peroxidation products (malondialdehyde and 4-hydroxynonenal, among others).74,75 They also present protein carbonylation secondary to oxidative stress76; these changes are not unique to PD, and have been observed in other neurodegenerative diseases.
Studies into mitochondrial DNA damage have contributed new hypotheses on the aetiology of some age-related neurodegenerative diseases. Several studies report mitochondrial complex I deficiency in the substantia nigra and prefrontal cortex of patients with PD, which suggests that these deficits are specific to this area.77,78 This particular change in complex I activity has also been described in the striatum and the substantia nigra in animal models and in patients with PD.79,80
Amyotrophic lateral sclerosis (ALS) is a fatal degenerative disease that affects motor neurons; it has been found to be associated with a mutation in the gene encoding Cu/Zn SOD, on chromosome 21; this alteration is only observed in familial cases of the disease, and follows an autosomal dominant inheritance pattern.81 This allows a more targeted approach to treatment not only of familial ALS but also of sporadic ALS, which accounts for most cases. The potential role of Cu/Zn SOD in ALS has been studied in transgenic mice overexpressing the human Cu/Zn SOD gene.82 The animals displayed pathological changes in motor neurons, including loss and destruction of axon terminals and the development of multiple small terminals.83 Toxicity secondary to increased SOD activity may be explained by the formation of H2O2, a precursor of OH radicals. OH radicals can react with a wide range of molecules, particularly polyunsaturated fatty acids, triggering lipid peroxidation and disrupting membrane integrity.84
Other findings demonstrate the important role of oxidative stress in the pathogenesis of both sporadic and familial ALS. The antioxidant activity of the onco-protein Bcl-2 can regulate ROS production during cell and neuronal death.85 Glutamate excitotoxicity has also been proposed as a pathophysiological mechanism in ALS.86,87 During this process, O2− may be generated in various ways, including direct ROS production, calcium-mediated xanthine oxidase activation, and initiation of the arachidonic acid cascade by phospholipase A2.88–90 Finally, NO, which is also produced in this way, can react with O2− to form ONOO−; these species are highly reactive.88
ConclusionsOzone air pollution induces a state of chronic oxidative stress, which plays a crucial role in the pathogenesis of chronic degenerative diseases, negatively affecting quality of life. Non-invasive animal models of chronic oxidative stress, replicating the current environmental conditions through exposure to low doses of ozone and inhibition of antioxidant defence systems, may help us to understand the association between oxidative stress and chronic degenerative diseases, providing relevant information about molecular and cellular interactions that affect different brain structures and induce neurodegeneration.
Chronic increases in ROS production, combined with dysfunction of the antioxidant defence system, represents a considerable risk of developing neurodegenerative diseases. Pro-oxidant molecules have a direct impact on synaptic plasticity, causing dendritic spine loss, neuronal death, and loss of the brain’s repair capacity. This results in loss of synaptic contacts, which affects information processing in neural networks. Increased ROS production also alters signalling cascades involved in gene expression, leading to cognitive deficits that may trigger neurodegenerative disease.
FundingThis study was funded by Support Programme for Research and Technological Innovation (PAPIIT-IN221417) and the Mexican National Council of Science and Technology (CONACyT 219703).
Conflicts of interestThe authors have no conflicts of interest to declare.
We would like to thank the General Directorate of Academic Personnel Affairs of the Universidad Nacional Autónoma de México for granting a doctoral scholarship to Paola Cristina Bello Medina.
Please cite this article as: Bello-Medina PC, Rodríguez-Martínez E, Prado-Alcalá RA, Rivas-Arancibia S. Contaminación por ozono, estrés oxidativo, plasticidad sináptica y neurodegeneración. Neurología. 2022;37:277–286.
