



Genetic analysis of specific language disorders is of major interest for both clinical research and linguistic theory. However, the results of this analysis almost always do not show any univocal and compulsory relationships between particular gene mutations and particular disorders or a causal link between the genotype and the phenotype.
ObjectivesThis paper will review this type of evidence (referring to the “language gene” FOXP2 as a leading example, where possible), try to suggest plausible reasons for such a perplexing output, and ultimately discuss if such reasons really explain the genuine aetiology of these conditions.
ResultsThe key to disentangle and understand the puzzling scenario emerging from the genetic analysis of specific language disorders is to pay attention to the actual role played by genes during ontogeny and, in particular, to the way in which developmental processes are actually regulated: genes are not direct causal agents regarding the emergence of impaired or wild phenotypes, but just one among the diverse types of regulatory factors involved.
ConclusionsWhen such a complex role as well as development models less focused on the genes are considered, the way in which genetic mutations really contribute to the emergence of these cognitive disorders is quite satisfactorily explained.
El análisis genético de los trastornos específicos del lenguaje resulta del máximo interés, tanto para la práctica clínica como para la teoría lingüística. No obstante, un resultado casi universal de dicho análisis es que no parece existir una relación unívoca y obligada entre la mutación de determinados genes y la aparición de patologías concretas al término del desarrollo, ni por consiguiente, una relación causal directa entre el genotipo y el fenotipo.
ObjetivosEl presente trabajo se plantea evaluar esta clase de evidencias (utilizando como modelo, allí donde resulte ilustrativo, el gen FOXP2, considerado habitualmente como el «gen del lenguaje» por excelencia), proponer posibles causas que expliquen su recurrencia y discutir si tales explicaciones contribuyen realmente a esclarecer la genuina etiología de estos trastornos.
ResultadosLa clave para entender el intrincado (y a primera vista desconcertante) escenario resultante del análisis genético de los trastornos específicos del lenguaje radica en atender al verdadero papel que desempeñan los genes durante la ontogenia y, especialmente, al modo en que se regulan los procesos de desarrollo: lejos de erigirse en los agentes causales directos responsables en exclusividad de la aparición de los fenotipos, los genes constituyen uno más de los múltiples factores implicados.
ConclusionesLa asunción de la complejidad de dicho papel, así como la conveniencia de considerar modelos alternativos del desarrollo, menos centrados en los genes (por paradójico que pueda parecer), permite explicar satisfactoriamente el modo en que las alteraciones génicas contribuyen a la aparición de este tipo de trastornos de la cognición.
The possibility of identifying the structural and functional nature of language-related genes—genes whose products presumably play an important role in regulating the development and function of the neural centres involved in processing linguistic stimuli—is a matter of utmost interest for two key reasons. On the one hand, it may serve to corroborate certain hypotheses put forth by linguistic experts who state that grammatical competence acquired by the individual at the end of the developmental stage (that is, acquired knowledge of his or her first language) cannot be regarded as the mere result of an inductive learning process based on exposure to information constituting linguistic input.1,2 In fact, those who support such innateness hypotheses, including Chomsky himself,1 have postulated that there may be a linguistic genotype consisting of all information necessary for language acquisition and not acquired by experience. This essentially refers to the building blocks of Universal Grammar.3 According to this model, language is acquired because linguistic input in the presence of that genotype promotes the appearance of the phenotype representing competence.3 On the other hand, the fact that many language disorders are hereditary4–6 seems to suggest that in order to reveal a disorder's true aetiology, we must first identify and characterise the mutations that have presumably affected one of the genes making up the genotype in question. With that in mind, a common working hypothesis in this field is that the anomalous and hereditary linguistic action pattern present in some subjects must be the result of mutations in specific genes. It goes on to state that these genes affect a specific area of competence and leave other cognitive capacities unaltered (although some may be involved in the action). This scenario is ultimately possible because the mutation of the genes in question creates structural and/or functional changes in the brain centres involved in language processing.
In recent years, researchers have identified and drawn up clinical profiles for a variety of syndromes, ailments, disorders and illnesses that are hereditary and only seem to affect language. These include specific language impairment (SLI: OMIM 602081); dyslexia (OMIM 127700), and speech sound disorder (SSD: OMIM 608445). They may also include a number of other disorders with much lower prevalence rates, such as Landau–Kleffner syndrome (OMIM 245570), rolandic epilepsy/sylvian seizures with verbal dyspraxia (OMIM 601085), and 22q13.3 deletion syndrome (OMIM 606232). Likewise, a number of genes have been identified as possible candidates or risk factors for the appearance of such disorders. Additionally, certain loci (physical sites on a chromosome) are linked to or associated with these disorders.4–6 Although it may seem paradoxical at first, the truth is that performing genetic analyses for language disorders has not contributed significantly to identifying their true aetiology. At the same time, this fact seems to suggest that genotype–phenotype relationships are not as direct as one might imagine.
In this study, we discuss some of the evidence supporting the above statement, providing data derived from the analysis of FOXP2 as our principal examples. The FOXP2 gene is normally considered to be one of the causal factors for specific language impairment, and also the predominant “language gene”.7–12 In light of this evidence, we will discuss whether or not re-examining the genotype–phenotype relationship is necessary in order to understand the true role of genes in the appearance of neurodevelopmental disorders. In the end, this will require us to relinquish overly gene-centric concepts of cerebral ontogeny.
ProcedureSeemingly problematic results from the genetic analysis of specific language disorders: FOXP2 as a paradigmThe results obtained from analysing genes linked to or associated with the different disorders specific to language are especially interesting.
- –
As a general rule, the degree of impairment in individuals presenting the same anomalous variant of any of the candidate genes varies considerably (variable penetrance). On some occasions, the disorder does not manifest (zero penetrance) or its signs are very slight (low penetrance). Consequently, individuals bearing the R553H mutation in the FOXP2 gene (the first identified mutation) will present symptoms that will differ somewhat.13,14 Added to this, researchers recently described a mutation (T1591C) that was completely asymptomatic in some members of the same family.15
- –
We often observe that different mutations of the same gene produce slightly different phenotypes, which may even be classified clinically as different disorders. These disorders may be specific to language, or they may be cognitive and affect language and other cognitive abilities simultaneously (under other circumstances, we may simply observe that the same locus is associated with different phenotypes or disorders). In the specific case of FOXP2, the disorder associated with most isolated mutations described to date (and most of the chromosomal translocation events that affect the gene sequence) has been described as orofacial dyspraxia linked to development, or as spastic dysarthria. However, there is no consensus as to the precise clinical category for the disorder and the true nature of the underlying deficit (speech disorder, language disorder, general cognitive disorder including linguistic deficits, motor disease affecting language).9,13,14,16 In any case, the most recently identified mutation (T1591C) produces a substantially different, more complex phenotype which includes focal epileptic episodes, cognitive deficit and a number of linguistic deficits.15 Furthermore, certain polymorphisms of the gene have been linked to entirely different cognitive disorders, such as schizophrenia17 or frontotemporal dementia,18 although appearance of these polymorphisms is not yet considered to be a causal or risk factor. It seems clear that different mutations of FOXP2 give rise to slightly different development processes in the cerebral areas in which the gene is expressed, and that the neurological changes resulting from those mutations can affect the intensity and nature of the clinical manifestations of a number of cognitive disorders. This could be due to their effects on language-related endophenotypes; one prime example is the modulating effect that rs1456031 TT and rs17137124 TT polymorphisms have on the way frontotemporal dementia manifests, due to their effect on verbal fluency.18 On top of all this, the cerebral areas mentioned above can hardly be considered as exclusively dedicated to the processing of linguistic stimuli. Changes in these areas caused by mutations in other genes tend to produce a variety of different deficits and disorders, including some which are not language-related. The primary pathology associated with the FOXP2 mutation seems to be located in the basal ganglia14,19; while their functions are largely motor and cognitive, they also include functions specifically related to language.20,21 At the same time, dysfunction in this subcortical structure due to mutations in genes other than FOXP2 gives rise to other disorders which are not exclusively language-related: Huntington disease,22 Parkinson disease,20,23 3-methylglutaconic aciduria,24 glutaric acidaemia type I,25 and progressive supranuclear palsy.26,27
- –
On the other hand, we must be aware that most genes whose mutation produces these disorders code for products that do not act alone; in order to be functional, the products must be integrated in multiprotein complexes. In the case of the FOXP2 protein sequence, the specificity of its bond to DNA (in terms of time and tissue type) seems to depend on or be facilitated by its interaction with other FOXP2 molecules (homodimerisation). It also has to do with the formation of heterodimers with FOXP1 or FOXP4 and its bond with the transcriptional co-repressor CtBP1.28,29 It is significant that mutations in genes that code for these proteins also seem to produce language and speech disorders, as shown by the case of FOXP1.30–32 However, drawing once more on FOXP1 as an example, deficits observed here are not exactly the same as those associated with mutation in FOXP2 (for example, mutation in FOXP1 would not cause verbal dyspraxia in most cases30,33). Rather, it would be more common for several different cognitive disorders to appear simultaneously (mutation in FOXP1 is associated with both intellectual disability32 and autism31).
- –
We also must be mindful of the fact that the products coded for by most of these genes normally act as part of a complex regulatory network.5 Another possibility, however, is that mutation in some target genes would fail to produce a phenotype similar to that caused by mutation in the regulator gene. Instead, it could provoke symptoms that might be identified clinically as belonging to another disorder. What is probably the most significant progress toward describing the genetic basis of language is currently being made at this level, in the regulatory network that specifically includes FOXP2. The recent decoding of a fraction of the components making up that network serves to confirm the important role genes play in modulating the development of brain centres involved in language processing.34–36 One FOXP2 target is CNTNAP2, which codes for a protein in the neurexin family that seems to play a part in regulating synaptogenesis37 and specifically participates in establishing the exclusive connectivity pattern in the frontal lobe.38 However, this gene appears in mutated form in individuals affected by a number of different neurological disorders, including SLI,39 various types of language delays and disorders40,41 and autism.42,43 Another of the FOXP2 targets is SRPX2, which codes for a protein with 3 sushi domains and an HYR domain. It may be involved in perinatal and postnatal maturation of specific circuits in the cerebral cortex (including those involved in speech control), considering the role it plays in the regulation of cell migration and adhesion.44,45 The SRPX2 gene is considered the most likely candidate for causing a type of rolandic (sylvian) epilepsy with verbal dyspraxia similar to that described by Scheffer et al,46 except for being linked to the X chromosome. This disorder is characterised by oral-motor apraxia and mild difficulties understanding certain language structures, among other symptoms.44 However, mutation of that gene also seems to cause a type of bilateral perisylvian polymicrogyria44. Characteristic symptoms of this disorder include dysarthria (or in some cases, absence of spoken language) in addition to mild intellectual disability.47 It has been suggested that rolandic (sylvian) epilepsy with verbal dyspraxia, and other types of benign childhood epilepsy with centrotemporal spikes (or rolandic seizures), may represent one end of a phenotypic continuum. Continuous spike-wave discharge during slow sleep (CSWDSS)44 would be on the same continuum, and Landau–Kleffner syndrome46 would be located at the other end. The most characteristic endophenotype of all of these disorders, CTS (focal, sharp diphasic centrotemporal spikes) is associated with the ELP4 gene.48 This gene codes for one of the components of the elongator protein complex, a multiprotein complex with histone acetylation activity that is involved in RNA transcription and tRNA modification. Its dysfunction has been linked to altered cell mobility and migration, which would specifically affect certain neuron populations during the development of the cerebral cortex.49 It is significant that the locus corresponding to the ELP4 gene has in turn been linked to SSD,50 which suggests that the comorbidity observed for certain types of rolandic epilepsy and SSD could be the result of pleiotropy in this gene. As for the rest, once the SRPX2 protein is secreted it interacts with a number of proteins, especially with PLAUR, which codes for the urokinase plasminogen activator receptor.51 Both SRPX2 and PLAUR are FOXP2 targets,15 and are particular in that in vitro, the R553H variant of the gene is unable to inhibit expression of both genes.15 To summarise, different genes which appear to belong to the same regulatory network (probably FOXP2, CNTNAP2, SRPX2, PLAUR and ELP4) are also linked to disorders that are markedly different in clinical terms (possibly SLI, SSD, autism, dyspraxia, apraxia, rolandic epilepsy, CSWDSS and Landau–Kleffner syndrome). Mutation of these genes causes a variety of different symptoms and endophenotypes which may or may not be language-related (language deficits, cognitive deficits, oral-motor disorders, CTS).
- –
In addition, a variety of different candidate genes and others considered as potential risk factors have been proposed for each of the specific language disorders. This is the case because the identity of these genes differs (up to a certain point) from one population to another, and/or according to the subtype in question. As a result, SLI may be caused by a mutation in the FOXP2 gene (although this aetiology is controversial because describing SLI as a sensorimotor disorder14,16 makes it incompatible with its own diagnostic criteria52,53). It might be better described as the cumulative effect of a number of less important genes.54 To date, different QTLs linked to or associated with SLI55,56 have been identified, and several candidate genes for the disorder have been proposed. Putting aside the previously discussed example of CNTNAP2, there may be two other products involved in regulating calcium metabolism: ATP13A4, which codes for a P5-type ATPase cation transporter,57 and ATP2C2, which codes for a transporter involved in translocating calcium and magnesium ions from the cytosol to the Golgi apparatus.58 Another candidate would be CMIP, which codes for a component in the molecular mechanism anchoring the cell membrane to the cytoskeleton. It may be active in the regulation of neuronal migration and/or formation of synaptic complexes.59 Significantly, both ATP2C2 and CMIP are associated with the phonological component of short-term working memory.60 Impairment of this component is a typical endophenotype (and probably the core disorder) in SLI,61 and also in dyslexia62 and SSD.63 While none of these genes has been linked to the regulatory network including FOXP2 to date, we know that in vitro, CMIP binds to FLNA, a phosphoprotein that may be active in the regulation of neuronal migration during embryogenesis.64 Mutation in the gene coding for that phosphoprotein may in some cases result in developmental disorders that specifically affect language.65
- –
Lastly, individuals affected by one of these disorders often display a normal candidate gene sequence (phenocopy).
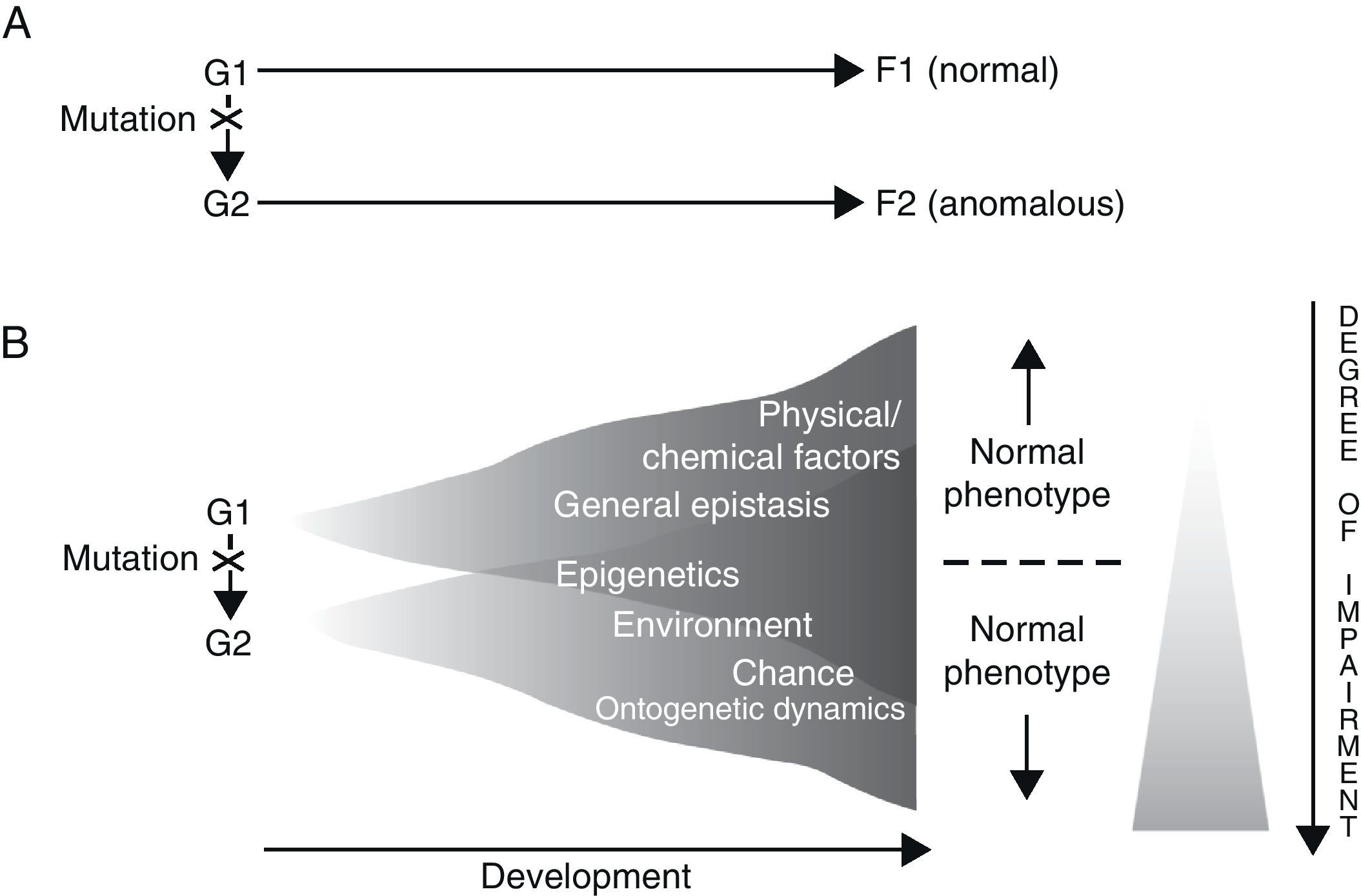
Results gathered from genetic analysis of specific language disorders, as described in the preceding section, seem to suggest at least two conclusions: on the one hand, for each disorder, there are a number of genes which may be considered as triggers or risk factors for the appearance of the disorder (polygenism), while at the same time, each gene fulfils separate functions in the organism at different locations and at different developmental moments (pleiotropism). For example, during embryonic development, FOXP2 is not only expressed in the brain, but also in the lungs, intestine and heart.66 On the other hand, a direct causal relationship between genotype and phenotype does not and cannot exist, for two fundamental reasons. The first is the intrinsic nature of genes and the way they function, and the second is the contribution of a long list of other, non-genetic factors to the regulation of development (Fig. 1). In fact, genes are not the primary cause of ontogenetic processes (whether normal or abnormal); rather, a gene is just one of the many factors that involved in a process, and in that process, each factor is equally necessary to the production of the final phenotype.67–69 We also must consider that development is always the result of synergistic interaction, and not a mere string of successive actions.70
 relationships with regard to language disorders. Adapted from Sholtis and Weiss.72")
Two alternative ways of envisioning genotype–phenotype (G–P) relationships with regard to language disorders. Adapted from Sholtis and Weiss.72
Numerous reasons support the above statement.
- –
To begin with, it is a mistake to think that genes are DNA sequences that intrinsically carry all of the information needed in order to synthesise a specific functional protein (or specific biochemical products, given that not all genes code for proteins71). On the contrary, most genes undergo post-transcriptional modifications and as such may generate alternative proteins (which are functionally different) based on the same primary transcript. They may even be generated by the combination of two different genes when chimeric transcripts form.73 In addition, many proteins must undergo post-translational modification (after synthesis) and/or join with other different proteins to form multiprotein complexes in order to be functional. Additionally, their action must occur at the right cell site, which means that they must be transported and translocated by the host of mechanisms regulating intracellular traffic. Once again, FOXP2 provides an enlightening example. This gene undergoes an alternative form of maturation in all species examined to date,74,75 and because of this, it gives rise to different protein isoforms which seem to play different physiological roles.76 However, as stated in the previous section, the FOXP2 protein seems to act within a multiprotein complex in which FOXP1, FOXP4 and CtBP1 (and possibly others) are also active. In the end, since at least the main isoform functions as a transcriptional factor, the protein must translocate to the nucleus in order to carry out that function. This process also requires the protein to have already formed bonds with other protein factors.28
- –
In addition, the degree to which a specific gene contributes to a specific biological process during development depends fundamentally on the time, location and quantity of synthesis of the product (or products) for which it codes. A number of extremely complex molecular mechanisms (cis-acting regulatory sequences, protein transcription and translation factors that are trans-acting, non-coding RNA [ncRNA] performing an intrinsic regulatory function or lending specificity to protein-type regulators) modulate these parameters with considerable precision and flexibility. This explains the pleiotropic nature of most of these genes, which was mentioned in the preceding section.
- –
On the other hand, in most cases (and especially in the case of regulator genes like FOXP2 [see above]), gene products do not act alone. In addition to forming specific protein complexes, these gene products tend to be integrated in complex regulatory networks.77 This also explains the polygenic nature of most of the phenotypic traits (recall that there are many genes in which mutation can have an effect on language, and also many candidate or risk factor genes for each specific disorder). Within these regulatory networks, the importance of the role played by each individual element is clearly secondary to the combined effect of all of the elements. At each given time and location, they form a precisely balanced mixture of all of the different products coded for by the set of genes involved. These products are usually arranged as gradients or specific combinations of signal molecules possessing physical and chemical properties that are especially relevant to regulating development processes (further explanation to follow). The homeostatic effect of the other components in the network and/or the presence of additional modifications at other network locations make it difficult to predict which phenotype is associated with the mutation of a specific gene, and phenotypes may vary considerably. (Recall that mutations in some of the FOXP2 targets led to cognitive disorders that were different from those associated with a mutation in the gene itself.) As a result, not even the presence of the same protein in the same location and at the same moment in development is enough to guarantee that the phenotype appearing under these conditions in a certain individual or population will also appear in another individual or population.
- –
Likewise, given that the cell in which a gene is expressed is not a closed system, gene expression will be subject to the continuous effect of a host of external factors. On the one hand, these factors will affect the other levels of complexity that can be defined for the neural substrates of language from a neurobiological standpoint (tissues, circuits, areas of the brain, etc.), but they ultimately come from the environment in which development occurs. These factors, whether endogenous or exogenous, do more than simply modify the genetic expression pattern through information transduction channels that activate regulatory cascades consisting of transcription and translation factors. In addition, they may also provoke epigenetic modifications in the genes themselves (that is, heritable alterations in DNA structure due to the methylation of certain bases or due to the modification of proteins responsible for organising them), and this also affects gene expression. Increasingly, epigenetic factors are considered to be a generalised and strategic mechanism for regulating cognitive and behavioural functions.78 We are increasingly led to believe that the transcriptional state of the cell—meaning which types of RNA are present, in what quantities and performing which functions—is especially important to the development and emergence of all types of phenotypic traits.71 Apart from epigenesis, other heritable factors exist which regulate an organism's development, especially during the early stages. Among these, protein gradients inherited from the maternal line are particularly important.79
- –
In addition, somatic variation may result in the appearance of genotypically different cell populations which express different varieties of the same gene (in the simplest scenario, a mutated gene and the wild-type gene). The influence of this phenomenon, associated with the activity of some kinds of transposable elements, can be especially significant during (and contribute to) nervous system development. It affects such important phenomena as neurogenesis and neural function, and ultimately, neural plasticity.80
- –
As for the rest, a number of general physical and chemical factors also play a decisive role in modulating development processes, and determine the ontogenetic route of all types of phenotypic traits (including viscoelasticity, biochemical diffusion and oscillation, the dynamic between sedimentation and diffusion gradients, mechanical and chemical excitability, and the dimensions of the space in which the chemical reactions take place. All of these factors act in conjunction with basic cell properties, such as polarity and differential adhesion81,82). These factors are crucial in determining the actions performed by all of the other regulatory elements involved (proteins, RNA, hormones, etc.), and they even explain basic aspects of the organisation of developing tissues, such as regionalisation and the appearance of morphological regularities.72,83
- –
Lastly, all development processes contain a highly stochastic component, which ultimately results in random interactions between the molecules involved. With this in mind, identical development processes that take place in equivalent environments may produce different phenotypes84; meanwhile, random selection also plays a key role in the evolution of the genotype–phenotype relationship.72 The importance of such stochastic effects has been shown to be especially important to brain development.84
In light of the above, it seems that the so-called “black box” concept of development85 is what ultimately explain how a single genotype can give rise to different phenotypes (phenotypic plasticity), and also how the same phenotype may be created by different genotypes (canalisation) (Fig. 1). By this logic, everything mentioned above should also enable us to interpret the perplexing scenarios revealed by genetic analysis of concrete language disorders, and therefore help us establish their true aetiologies.
Since pleiotropy is a given, the mutation of a certain gene may affect the normal development of 2 (or more) different brain structures. This results in structural and/or functional anomalies in 2 (or more) different neural components, which translates to the appearance of 2 (or more) different processing deficits. In turn, these deficits may manifest as a number of different symptoms on the phenotypic level. These symptoms are likely to be categorised clinically as belonging to 2 (or more) different disorders, which would be heterogeneous in some cases and comorbid in others. However, since our context is also polygenic, it may happen that the mutation of 2 (or more) functionally related genes in some cases would affect the development of the same brain structure. Similar structural and functional anomalies would therefore be present in the same neural component, producing a single processing deficit. In this case, we would detect the presence of similar symptoms on the phenotypic level, and they might be categorised clinically as corresponding to a single disorder, which could also be heterogeneous. However, another possibility—occurring in the concrete case of FOXP2 and its targets—is that the mutation of 2 (or more) such functionally related genes would affect the normal development of 2 (or more) different brain structures. As a result, structural and functional anomalies present in this case would affect 2 (or more) different neural components, producing 2 (or more) different processing deficits. On a phenotypic level, this would produce a number of symptoms likely to be categorised clinically as belonging to 2 (or more) different disorders which in some cases would be heterogeneous, and in others, comorbid. Lastly, it is important to recall that each dysfunctional or non-functional product's contribution to the production of an anomalous phenotype always depends on the subtle effects of all other genes involved, and on other development modulation factors in play (epigenetic, maternal, ontogenetic, environmental, etc.). As a result, mutations occurring in the same gene in different individuals or populations may result in different degrees of impairment having to do with the structural and functional integrity of specific areas of the brain. They would therefore produce variable processing deficits and cognitive profiles, in addition to a range of different symptoms. The latter are likely to be categorised clinically as 2 (or more) subtypes of the same disorder, or even 2 (or more) different disorders, which would in some cases be comorbid. For similar reasons, the reverse can also occur: the mutation of 2 (or more) different genes can, in certain individuals or populations, give rise to similar structural and functional anomalies in the same area of the brain (or in multiple areas). As a result, processing deficits and cognitive profiles would be similar, and ultimately give rise to symptoms likely to be categorised clinically as part the same disorder, or as different subtypes of the same disorder.
FundingThis study was completed as part of the research project Biolinguística: evolución, desarrollo y fósiles del lenguaje [Biolinguistics: evolution, development and fossils of language] (FFI2010-14955/FILO), supported by the Ministry of Education and Science and partially funded by FEDER.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Benítez-Burraco A. Aspectos problemáticos del análisis genético de los trastornos específicos del lenguaje: FOXP2 como paradigma. Neurología. 2012; 27:225-33.
