




A thorough knowledge of the socioeconomic scope of neuromuscular disease is essential for managing resources and raising social awareness.
DevelopmentOur group reviewed current data on the epidemiology, mortality and dependence rates, and socioeconomic impact of amyotrophic lateral sclerosis and neuromuscular diseases in Spain. We also recorded how neurological care for these patients is organised.
ConclusionsNeuromuscular disorders are a very heterogeneous group of diseases, and some are very rare. These disorders account for between 2.8% and 18% of the total motives for a neurological consultation. In Spain, prevalence and incidence figures for amyotrophic lateral sclerosis are similar to those in other countries; however, figures for patients with other neuromuscular diseases are not known. Since the diseases are chronic, progressive, and debilitating, they cause considerable disability and dependence, which in turn directly affects healthcare and social costs associated with the disease. The costs generated by one patient with amyotrophic lateral sclerosis or Duchenne disease have been calculated at about 50000 euros per year. Neuromuscular disease shows aetiological, diagnostic, and prognostic complexity, and it requires multidisciplinary management. Follow-up for these patients should be entrusted to specialised units.
El conocimiento del alcance socioeconómico de la patología neuromuscular es esencial para la planificación de recursos y la concienciación social.
DesarrolloSe ha realizado una revisión de los datos publicados hasta el momento sobre epidemiología, mortalidad, dependencia e impacto sociosanitario de la esclerosis lateral amiotrófica y las enfermedades neuromusculares en España. Además, se ha recogido cómo está organizada la atención neurológica en estos pacientes.
ConclusionesLa patología neuromuscular constituye un grupo muy heterogéneo de enfermedades, algunas de las cuales se consideran raras por su baja frecuencia. Esta patología supone entre el 2,8 y el 18% de los motivos de consulta en un Servicio de Neurología. En España, las cifras de prevalencia e incidencia de esclerosis lateral amiotrófica son similares a otros países; sin embargo, se desconoce el número de pacientes con otras enfermedades neuromusculares. Son enfermedades crónicas, progresivas y debilitantes, lo que condiciona una importante discapacidad y dependencia. Esto repercute directamente en los costes sanitarios y sociales asociados a la enfermedad. Se ha calculado que el coste de un paciente con esclerosis lateral amiotrófica o enfermedad de Duchenne se acerca a los 50.000 euros anuales. La patología neuromuscular tiene una gran complejidad etiológica, diagnóstica y pronóstica, y requiere un manejo multidisciplinar. Las Unidades especializadas deben ser las encargadas del seguimiento de estos pacientes.
The purpose of the Foundation for the Brain is to raise awareness of the impact of neurological diseases on patients and their families, friends, and carers, with the ultimate goal of improving patients’ and relatives’ quality of life. To this end, the Foundation for the Brain has developed a series of initiatives in the fields of information, prevention, research, patient care, and social integration. As part of these activities, the Foundation for the Brain prepares reports on the societal impact of different neurological diseases in Spain.
This report focuses on neuromuscular disorders (NMD), a large, complex group of disorders that merit separate analysis in view of their individual characteristics. More specifically, this report addresses amyotrophic lateral sclerosis (ALS) and other relevant non-ALS neuromuscular disorders.
Neuromuscular disorders affect the peripheral nervous system, skeletal muscle, neuromuscular junctions, and spinal cord. This heterogeneous group includes a wide range of entities (Table 1), classified by lesion location: motor neuron diseases, radiculopathies, plexopathies, neuropathies, neuromuscular junction diseases, and muscle disorders. These conditions may be metabolic, infectious, toxic, immune-mediated, genetic, or neurodegenerative in origin. They may manifest during childhood or adulthood and may be acute, subacute, or chronic. NMDs require highly specialised diagnosis and treatment; the study and management of these disorders therefore constitutes a subspecialty within general neurology.
Main neuromuscular disorders by location.
| Inherited | Acquired | |
|---|---|---|
| Motor neuron diseases | Spinal muscular atrophy Hereditary spastic paraplegia | Amyotrophic lateral sclerosis Poliomyelitis |
| Peripheral nerve diseases | Charcot-Marie-Tooth disease Hereditary neuropathy with liability to pressure palsies Hereditary sensory and autonomic neuropathies Distal hereditary motor neuropathies | Guillain-Barré syndrome Chronic inflammatory demyelinating polyneuropathy Metabolic neuropathies Toxic neuropathies Nutritional neuropathies Paraneoplastic neuropathies |
| Neuromuscular junction diseases | Congenital myasthenic syndrome | Myasthenia gravis Eaton-Lambert syndrome Botulism |
| Muscle disorders | Muscular dystrophies -Myotonic dystrophy -Facioscapulohumeral muscular dystrophy -Dystrophinopathies (DMD, BMD) -Limb-girdle muscular dystrophy -Congenital muscular dystrophies Congenital myopathies Distal myopathies Mitochondrial myopathies Metabolic myopathies | Inflammatory myopathies Viral myositis Toxic neuropathies Myopathies secondary to endocrine disorders |
BMD: Becker muscular dystrophy; DMD: Duchenne muscular dystrophy.
ALS, including its variants (primary lateral sclerosis, progressive muscular atrophy, and progressive bulbar palsy), is the most frequent motor neuron disease in adults. This disabling, progressive neurodegenerative condition affects both the upper (located in the precentral motor cortex) and the lower motor neurons (located in the motor nuclei of the brainstem and the anterior horn of the spinal cord). Although it is typically sporadic (an isolated case in a family), 5% to 10% of cases are clustered in families, most frequently following an autosomal dominant inheritance pattern (however, autosomal recessive and X-linked dominant inheritance patterns have also been reported). Around 5% to 10% of cases also exhibit dementia, normally frontotemporal dementia, which may appear before, after, or simultaneously with ALS onset. However, neuropsychological tests have found evidence of executive dysfunction in over 50% of patients with ALS.
ALS is a progressive, invariably fatal disease. Patients typically die within 5 years of onset as a consequence of progressive restrictive respiratory failure. There is still no curative treatment for ALS; current treatment approaches focus on providing palliative care to control symptoms from disease onset.
Though diverse, non-ALS NMDs share certain characteristic features with similar consequences, which explains why they are grouped together. The most common clinical manifestation of these disorders is loss of strength, which is usually progressive. Muscle weakness may cause a number of orthopaedic disorders, including rigidity and joint deformities, which are especially pronounced in paediatric patients. Other frequent motor symptoms include fatigue, contractures, or difficulty relaxing muscles. The main non-motor manifestations of NMDs are sensory alterations, pain, and dysautonomia. Respiratory and/or cardiac disorders may appear as the disease progresses; these problems constitute the main cause of death in patients with non-ALS NMDs. These patients may also experience swallowing dysfunction, necessitating gastrostomy feeding. Non-ALS NMDs must therefore be managed by a multidisciplinary team.
Some NMDs are frequent; acquired polyneuropathies, for example, affect 8% of the general population. Other NMDs, however, are considered to be rare diseases, with a prevalence below 5 cases per 10000 population in the European Union (Regulation [EC] No. 141/2000 of the European Parliament). Although the individual prevalence of each disorder in the group is low, the overall prevalence of NMDs is considerable. In Spain, approximately 3 million people have rare diseases. Most rare NMDs are genetically determined and onset may occur during childhood. Several types of symptomatic treatment are available; however, most of these disorders have no curative treatment, and their course is therefore chronic, progressive, and severely disabling. Given their low prevalence, these conditions are little understood by society in general or by the medical community, leading to undesirable diagnostic delays. In Spain, epidemiological and pathophysiological research into these entities has been promoted in recent years through the activity of the Spanish Society of Neurology's Neuromuscular Disorders Study Group, the development of the Orphanet portal, and the creation of the Biomedical Research Network for Rare Diseases (CIBERER). CIBERER has developed several genetic medicine research programmes focusing on such NMDs as muscular dystrophy, spinal muscular atrophy, and Charcot-Marie-Tooth disease (CMTD). In view of the great personal and social impact of rare NMDs, our report will focus on these conditions.
EpidemiologyEpidemiology of amyotrophic lateral sclerosisALS is the third most frequent neurodegenerative disease, after dementia and Parkinson's disease. Extensive research into the epidemiological characteristics of ALS has included studies on the general population, specialised units, and geographical areas with a high incidence of the disease. Studies on Western populations usually report an incidence of 1 to 2 cases per 100000 person-years, except for populations in high-incidence regions (Guam, Kii peninsula, Irian Jaya, a tribe in Angurugu [Australia], and Guadeloupe in the Caribbean). The literature also provides data about the incidence and prevalence of ALS in different ethnicities. Studies on ethnically mixed populations have found higher incidence of ALS in white people than in multiracial people,1 and lower incidence in African Americans.2 ALS is estimated to affect one in 400 to 800 people, if they live long enough.
Prevalence of ALS (which is dependent on survival, and is therefore affected by the development of new treatments) is low due to high mortality rates; prevalence ranges between 2 and 5 cases per 100000 population (although higher and lower rates have been reported).
Few epidemiological studies of ALS have been conducted in the Spanish population. These have focused on the regions of Cantabria,3 Segovia,4 the island of La Palma (in the Canary Islands),5 and more recently Catalonia (with higher methodological quality).6 The former studies report somewhat lower incidence rates; the latter study, however, reports similar incidence and prevalence rates to those of other studies (1.4 and 5.4 cases per 100000 population, respectively).
Genetic causes are also universally distributed. Nonetheless, some mutations are more frequent in specific areas, which may be explained by the founder effect (C9orf72 expansion in Sardinia or Finland, recessive ALS associated with the p.D90A mutation in SOD1 in Sweden, and the p.A4V mutation in SOD1 in the United States). Mean age at onset ranges from 60 to 69 years; peak incidence occurs between the ages of 70 and 75 and decreases thereafter, unlike in Parkinson's disease or Alzheimer disease. All series of Western populations have reported a slightly higher incidence of ALS in men than in women, whereas studies including African American populations report the opposite effect. In terms of location of onset, ALS is bulbar in nearly one third of patients and spinal in the remaining 2 thirds (affecting the upper and lower limbs equally). ALS has a generalised or respiratory onset in a small number of cases.
Based on the above, ALS is estimated to affect 3 new patients per day in Spain; at present, there are over 3000 ALS patients in Spain. Given that the peak incidence of ALS is slightly lower than that of other neurodegenerative diseases, over 50% of cases would correspond to fully productive, working-age individuals.
Epidemiology of neuromuscular disordersNMDs are difficult to study as a whole due to the wide range of conditions included in this group; furthermore, few epidemiological studies have included all or most NMDs. Emery's7 1991 study continues to be a major reference. The author reviewed over 150 publications on the frequency of multiple hereditary NMDs affecting children and adults, including muscular dystrophy (Duchenne muscular dystrophy [DMD], Becker muscular dystrophy [BMD], limb-girdle muscular dystrophy, and facioscapulohumeral muscular dystrophy [FSHMD]), myotonic disorders (Steinert disease and myotonia congenita), spinal muscular atrophy, and hereditary sensorimotor neuropathies. Emery estimated global prevalence of NMDs at 286 cases per million population; one in every 3500 people may therefore develop a disabling NMD at some point in their life. During the 1990s, several population-based epidemiological studies were published in Europe. In Örebro, Sweden, the prevalence of NMDs (including motor neuron diseases, hereditary neuropathies, myotonic disorders, muscular dystrophies, and myositis) was estimated at 84 cases per 100000 population.8 In Northern Ireland, an analysis including hereditary NMDs only estimated prevalence at 34.5 cases per 100000 population (1 case in every 2900 people).9 Regarding paediatric populations, the prevalence of NMDs in patients younger than 20 years was 24.9 cases per 100000 population in Bologna.10 In western Sweden, however, prevalence was estimated at 63.1 cases per 100000 population for all NMDs and 53.1 cases per 100000 population for hereditary NMDs only.11
The prevalence of all NMDs in Spain is unknown. According to data from ASEM, a federation of 21 Spanish associations of patients with NMDs, over 60000 people in Spain have NMDs.
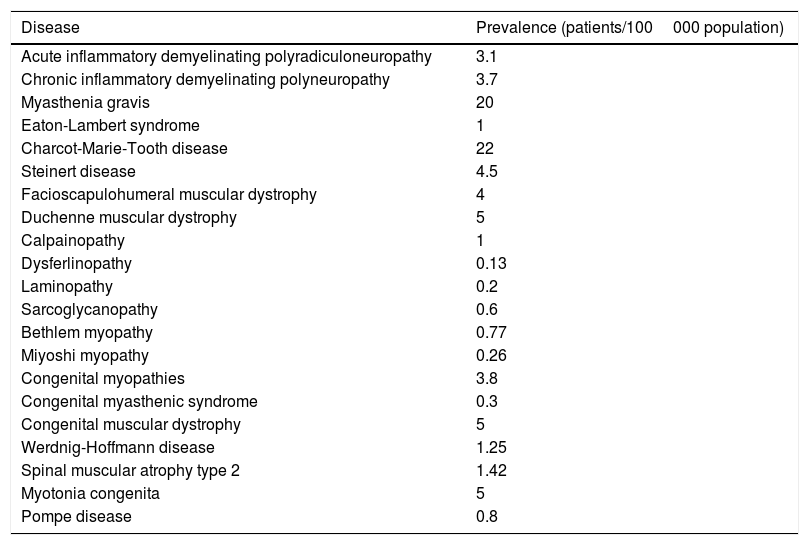
More data are available from individual analyses of each NMD. Orphanet is the leading European web portal for rare disorders and orphan drugs. In May 2014, Orphanet published estimated prevalence rates for rare diseases in Europe, based on the data gathered from a systematic literature search on Medline, multiple websites, patient registries, and medical reports.12 Prevalence rates for each NMD are shown in Table 2. Although extremely valuable, this study has several limitations. Firstly, the authors acknowledge the low level of consistency between sources and occasional confusion between prevalence and incidence. Furthermore, the reported rates may be overestimated, given that many studies are conducted in hospital settings or in geographical areas with a high prevalence of a given disorder. Surprisingly, the list of NMDs analysed by Orphanet did not include BMD or McArdle disease.
Estimated prevalence of NMDs (Orphanet Report Series, May 2014).
| Disease | Prevalence (patients/100000 population) |
|---|---|
| Acute inflammatory demyelinating polyradiculoneuropathy | 3.1 |
| Chronic inflammatory demyelinating polyneuropathy | 3.7 |
| Myasthenia gravis | 20 |
| Eaton-Lambert syndrome | 1 |
| Charcot-Marie-Tooth disease | 22 |
| Steinert disease | 4.5 |
| Facioscapulohumeral muscular dystrophy | 4 |
| Duchenne muscular dystrophy | 5 |
| Calpainopathy | 1 |
| Dysferlinopathy | 0.13 |
| Laminopathy | 0.2 |
| Sarcoglycanopathy | 0.6 |
| Bethlem myopathy | 0.77 |
| Miyoshi myopathy | 0.26 |
| Congenital myopathies | 3.8 |
| Congenital myasthenic syndrome | 0.3 |
| Congenital muscular dystrophy | 5 |
| Werdnig-Hoffmann disease | 1.25 |
| Spinal muscular atrophy type 2 | 1.42 |
| Myotonia congenita | 5 |
| Pompe disease | 0.8 |
Although we were unable to analyse each NMD separately, the present report does analyse the more representative NMDs more thoroughly: the 3 most frequent muscular dystrophies (Steinert disease, FSHMD, and DMD), CMTD, Guillain-Barré syndrome (GBS), and myasthenia gravis (MG).
Myotonic dystrophy. Steinert diseaseMyotonic dystrophy (MD) is the most frequent type of muscular dystrophy in adults, and follows an autosomal dominant inheritance pattern. This multisystem disorder is characterised by muscle weakness and myotonia, cataracts, heart disease, gastrointestinal problems, and hormonal alterations.13 Worldwide prevalence of Steinert disease ranges from 2.1 to 14.3 cases per 100000 population, depending on the series. The highest prevalence rate was reported in Saguenay-Lac-Saint-Jean, in the region of Quebec, with 158 cases per 100000 population in 2010.14 In the early 1990s, 2 studies on the epidemiology of MD were published in Spain. Based on hospital medical records, Burcet et al.15 estimated the prevalence of MD on the island of Mallorca at 11 cases per 100000 population. Soon after, López de Munain et al.16 reported a prevalence of 26.5 cases per 100000 population in the Spanish province of Guipúzcoa. This is one of the highest rates reported worldwide; according to the authors of the study, this may be explained by methodological factors (comprehensive search, use of different sources) and possibly a local founder effect, as in Saguenay-Lac-Saint-Jean.
Facioscapulohumeral muscular dystrophyFSHMD is characterised by asymmetric muscle weakness, predominantly affecting the muscles of the face and the shoulder girdles. It has an autosomal dominant inheritance pattern. After MD, FSHMD is one of the most frequent types of muscular dystrophy. In his 1991 study, Emery reported prevalence rates between 2.2 and 66.9 cases per million people in Western countries.7 A population study conducted in the province of Padua found a prevalence of 44 cases per million people, providing more recent data from another Mediterranean country similar to Spain.17
No data are available on the prevalence of FSHMD in Spain. The survey on disability, personal autonomy, and dependency status conducted in Spain in 2008 did not include FSHMD as a distinct entity, although it did include a group labelled “muscular dystrophies”, with 138 200 patients aged 6 to 64 years.18
DystrophinopathiesDystrophinopathies are significant among NMDs. They follow an X-linked recessive inheritance pattern. Genetic alterations causing a total absence of dystrophin cause DMD, whereas those which leave a reduced amount of dystrophin cause BMD. DMD has traditionally been considered the most frequent genetic muscle disorder in childhood, with an estimated incidence of 1 case per 3500 boys.7 Incidence of BMD is lower, at 5.4 cases per 100000 population, with a prevalence of 2.8 cases per 100000 population. In 2014, Mah et al.19 published a meta-analysis on the epidemiology of dystrophinopathies. The paper reviewed 31 studies, with all the population-based studies included in the meta-analysis having been conducted in North America and Europe. Prevalence rates for DMD and BMD were estimated at 4.78 and 1.53 cases per 100000 male patients, respectively. Incidence of these conditions could not be analysed due to the low number of studies conducted and the disparity of the data. In the case of DMD, incidence ranged between 10.71 cases (in Italy) and 27.78 cases (in Canada) per 100000 male live births per year.
No data on the prevalence or incidence of these conditions are available for Spain, except for generic data on “muscle dystrophies” provided by the 2008 survey on disability, personal autonomy, and dependency status mentioned above.18
Charcot-Marie-Tooth diseaseCMTD is a group of genetically heterogeneous hereditary neuropathies affecting motor and sensory function. CMTD is one of the most common hereditary NMDs, with a prevalence of as many as 40 cases per 100000 population.20 A pioneering study in Spain prospectively analysed the frequency of CMTD in the region of Cantabria between the years 1974 and 1984. At that time, CMTD was diagnosed based on clinical manifestations, the inheritance pattern, and neurophysiological findings. Prevalence in 1985 was 28.2 cases per 100000 population.21 More recently, several descriptive studies have been published, including series with very large numbers of Spanish patients. Although these are not population-based studies, they are relevant in that they underline the clinical and genetic characteristics of patients with CMTD in a specific region.22,23
Guillain-Barré syndromeGBS is an acute inflammatory immune-mediated polyradiculoneuropathy. Unlike most polyneuropathies, GBS develops rapidly. A meta-analysis of the incidence of GBS was conducted in 2011. This study reviewed all articles published between 1966 and 2009, and finally included 16, reporting an annual incidence of 1.11 cases per 100000 population (range, 0.81-1.89).24 An important study in the Spanish region of Cantabria showed an annual incidence of one case per 100000 population between 1975 and 1988.25 The growing interest in GBS surveillance in Spain led to the creation of the GBS Epidemiological Study Group, with 11 participating centres. This study group retrospectively gathered all patients with GBS aged 20 and older between 1985 and 1997, reporting an incidence of 0.85 cases per 100000 person-years. This rate was found to increase with age and time.26 As of 1996, the group's analysis became prospective, with a minimum follow-up period of one year. Between 1998 and 1999, incidence increased to 1.26 cases per 100000 person-years; this rate is similar to those found in other western European countries.27
Myasthenia gravisMG is an autoimmune disease of the neuromuscular junction, characterised by muscle weakness and fatigue. A 2010 meta-analysis of 55 studies published between 1950 and 2007 reported an incidence of 5.3 cases per year per million population (range, 1.7-21.3) and a prevalence of 77.7 cases per million population (range, 15-179).28 Incidence and prevalence have been found to increase over the years due to biological factors, better patient identification methods, and a change in the natural history of the disease due to treatment advances. Two population-based epidemiological studies of MG have been conducted in Spain. In the first of these, which was conducted on the island of La Palma, prevalence of MG was estimated at 8.58 cases per 100000 population in 1996.29 The second paper was a prospective study (1991-2000) performed in the area of Osona, in the province of Barcelona, and reported an annual incidence of 21.27 cases per million population. Interestingly, the annual incidence of MG increased with age, reaching 63.38 cases per million people in the population aged over 65 years. This would support the hypothesis of an unexpected increase in autoimmunity in the elderly.30
As mentioned previously, few data are available on the total number of patients with NMDs and the frequency of each of these disorders in Spain; developing a Spanish national patient registry is therefore essential. To address this, a project was launched in 2010 to create a database including all patients with NMDs in Spain. The project included a group of researchers specialising in NMDs from 7 Spanish autonomous communities and belonging to 6 CIBERNED research groups, one CIBERER research group, and 6 associated research groups. The purpose of the registry was to provide comprehensive, unified data for Spain and to analyse the distribution of each NMD, which is essential for healthcare resource planning. These databases may be useful for developing clinical and basic research initiatives, unifying and validating diagnostic methods, designing diagnostic and therapeutic algorithms, and conducting prospective treatment trials. This complex registry is based on a model implemented in the Netherlands, which successfully established a flow of information between NMD specialists and resulted in a number of excellent studies. At present, the Neuromuscular Disorders Database has gathered data on over 5000 patients with different NMDs, providing data for multicentre studies of the most prevalent disorders, such as ALS.31 The Neuromuscular Disorders Database forms part of the international TREAT-NMD Neuromuscular Network. This has resulted in better representation of Spanish patients with NMDs in European epidemiological and clinical projects.32
MortalityMortality associated with amyotrophic lateral sclerosisALS progresses rapidly and has a fatal course. Nearly half of patients die within 3 years of disease onset, 80% within 5 years, and over 95% within 10 years. According to a study conducted in Spain analysing mortality data for the period between 1951 and 1990, mortality due to ALS has increased over the years. This study reported an overall mortality rate of 1.49 per 100000 population, with a slightly higher rate in men (1.90 vs 1.21 in women); mortality peaks between the ages of 60 and 69.33
Mortality associated with neuromuscular disordersLife expectancy associated with most NMDs is very short; patients may die during childhood. Ventilatory failure and heart disease (structural or arrhythmogenic heart disease) are the main causes of death.
DMD is a paradigmatic NMD due to its fatal course. Its natural history has changed in recent years thanks to the early provision of multidisciplinary treatment combining corticosteroids and good management of respiratory, cardiac, nutritional, and orthopaedic complications. Historical series report that patients died soon after adolescence; however, survival has increased to the third, and exceptionally the fourth, decade of life with the introduction of non-invasive ventilation and the systematic approach to spinal surgery.34
While some NMDs follow a chronic course, other entities are susceptible to rapid change or potentially severe exacerbations. This is the case with MG and GBS. Myasthenic crises, the most severe clinical situation in MG, are characterised by acute respiratory failure requiring mechanical ventilation. Fifty years ago, mortality associated with myasthenic crises surpassed 30%.35 At present, however, mortality rates have decreased considerably, due to the introduction of immunomodulatory therapy for long-term management and intensive specialised care for acute complications. This has resulted in an increase in the prevalence of MG. The previously mentioned meta-analysis of MG epidemiology included 7 studies providing mortality data; mortality rates in these studies ranged from 0.06 to 0.89 deaths per million people per year.28 A retrospective study conducted by Alshekhlee et al.36 in the United States calculated in-hospital mortality in patients with MG between 2000 and 2005. The study included data from 5502 patients admitted to different centres; 2.2% of these patients died during hospitalisation, and 4.47% during myasthenic crises. Predictors of death were older age and respiratory failure. Mortality due to GBS has traditionally been estimated at 1% to 5% of patients, and is linked to respiratory and/or cardiovascular complications. Alshekhlee et al.37 published another study on in-hospital mortality due to GBS between 2000 and 2004 using a similar methodology to that of their study on MG. The study included a total of 4954 patients, 2.58% of whom died during hospitalisation, with no significant changes in mortality rates over the years. The main predictor of death was orotracheal intubation.
Spain's National Statistics Institute provides data on the rates and causes of mortality for multiple conditions. For the data to be included in the National Statistics Institute database, death certificates must indicate diagnosis of the disease (in this case, NMDs) using the ICD-10 classification. This requirement, however, is not always met. Mortality by cause of death for the year 2012 is listed as absolute values (mortality rates are not provided), as follows: myotonic disorders, 134; muscular dystrophies, 101; myasthenia gravis, 91; Guillain-Barré syndrome, 45; and hereditary motor and sensory neuropathy, 6.38
Disability and dependenceDisability is defined as a functional limitation in the activities of daily living due any type of deficiency; dependence is a consequence of disability. In the case of NMDs, deficiencies include weakness, joint contractures, scoliosis, cardiopulmonary dysfunction, pain, and cognitive impairment, and lead to decreased mobility, difficulty performing the activities of daily living, increased fatigue, cognitive/learning disorders, and poor psychosocial adaptation. Disability results in fewer educational and employment opportunities, greater dependence, and poorer quality of life.39,40 Dependence involves the need for assistance or care; the figure of the carer is therefore of great importance for these patients. In the case of patients developing an NMD during childhood or adolescence, the family and societal burden will persist throughout the patient's life.
According to the Survey on Disability and Dependence conducted in Spain in 2008, MD and ALS are among the main causes of disability. These 2 entities are the only NMDs addressed by the survey, which does list other recognised, homogeneous neurological diseases, such as Parkinson's disease, cerebrovascular diseases, Alzheimer disease, other dementias, multiple sclerosis, acquired brain damage, spinal cord injury, autism, Down syndrome, and cerebral palsy.18
Disability and dependence in amyotrophic lateral sclerosisALS, a debilitating condition progressively leading to complete paralysis, is an archetypal model of a disease causing high levels of disability and dependence. Patients with ALS rapidly progress from normal function to complete dependence. The impact of disease progression on functional ability is such that the main tool for measuring disease progression and assessing treatment effectiveness is a scale that evaluates disability (ALSFRS-R). This scale rates disease progression based on the patient's limitations in 4 functional areas: bulbar function, fine motor tasks, gross motor tasks, and respiratory function. Patients with ALS will experience some type of disability from disease onset, which will progressively increase with the course of the disease.
Disability and dependence in neuromuscular disordersMotor alterations are the most severe and evident limitations in NMDs. Several clinical scales have been developed to evaluate disease progression. Some of these can be applied to any type of NMD (Motor Function Measure, Neuromuscular Score, Brooke scale, Vignos scale, and even some items of the ICF), whereas others are designed to assess a specific condition; examples are the NorthStar Ambulatory Assessment (for DMD) and the Hammersmith Functional Motor Scale (spinal muscular atrophy).41,42 Such non-motor deficiencies as pain or cognitive impairment also have a considerable functional impact. Chronic pain is a significant element of the burden of disease associated with NMDs.43,44 Although neuropathic pain is well documented in patients with neuropathies, many NMDs are associated with different types of pain. Guy-Coichard et al.45 analysed the presence of chronic pain in patients with different NMDs (DMD, MD, FSHMD, metabolic myopathies, and MG) with a questionnaire combining different specific scales. A total of 511 patients responded to the survey, 331 of whom (67.3%) reported having experienced pain in the previous 3 months. Pain intensity varied between entities and was most severe in patients with metabolic myopathies and MG.
Cognitive impairment is present in some NMDs, including DMD, MD, and congenital muscular dystrophies associated with brain malformations. The associated disability is similar to that caused by other neurological diseases affecting intellectual function; in the case of NMDs, however, cognitive impairment further affects overall disability, increasing the degree of dependence.
Economic impactDisability and dependence associated with NMDs have a significant impact on society and on healthcare systems. The chronic, debilitating nature of ALS and most NMDs results in a great economic burden on families and on the state. Patients must undergo regular follow-up assessments and physiotherapy sessions and may at times need to be admitted to hospital due to exacerbations or intercurrent processes. These patients usually require technical support and orthotic/prosthetic materials which are not always covered by the Spanish social security system. Many patients also need assistance from a carer to complete the activities of daily living, and frequently require adapted housing and transportation. NMDs also cause significant changes to patients’ and carers’ working lives, leading to decreased income.
The global costs of NMDs involve both direct and indirect costs. Direct costs include medical care, outpatient care, and treatments, whereas indirect costs are those associated with patients’ or carers’ decreased productivity as a result of inability to attend work or early mortality. There are also intangible costs associated with patients’ suffering due to their decreased quality of life.
Economic impact of amyotrophic lateral sclerosisNo studies measuring the economic impact of ALS have been conducted in Spain; however, several studies addressing this topic have recently been published in other countries. For example, a study conducted in Ireland estimated the monthly cost of managing ALS from diagnosis to death at €1795.21 per patient; 21% of these costs were attributed to the use of multidisciplinary units, 72% to community-based care, and 7% to aids and appliances.46 Higher costs were associated with patients with rapid disease progression and those undergoing gastrostomy or receiving non-invasive mechanical ventilation. The costs associated with informal care and loss of productivity were not analysed in the study.
A study conducted in Canada estimated the annual direct costs per patient of ALS management at $32337, $19574 of which was paid out-of-pocket by patients or their families. The most significant direct costs were related to home adaptations, mobility aids, medical expenses, and private personal support workers. The study also estimated indirect costs associated with decreased income at $56821 per year.47 A similar study conducted in the United States estimated total costs over the course of the disease at $1433992 per patient; 85% was paid by insurance companies, 9% by families, and 6% by charities. The highest costs were associated with the need for at-home carers ($669150), mechanical ventilation ($212430), and hospital care ($114558).48 A study conducted in the Netherlands compared the monthly costs of care for a group of patients cared for in specific multidisciplinary units and a group of patients receiving general care. Interestingly, differences between the groups were found to be negligible (monthly cost of €1336 for multidisciplinary units vs €1271 for general care).49
Lastly, in a recently published study from the United States, Larkindale et al.50 estimated annual per-patient costs of ALS, DMD, and MD at $63693, $50952, and $32236, respectively. According to this study, annual costs of ALS in the US were $1023 million vs $787 million for DMD and $448 million for MD.
Treatment decision-making is therefore greatly influenced by the high economic burden associated with the management of these conditions and the possibility of covering these costs.
Economic impact of neuromuscular disordersStudies addressing the economic impact of NMDs are scarce, and mainly focus on the most disabling types, such as muscular dystrophies, especially DMD. Ouyang et al.51 studied the costs of managing muscular dystrophy (subtypes were not specified) in the United States in 2004. The study included only privately insured patients who were younger than 30. Annual medical costs were estimated at $20467 per patient. However, costs amounted to $34161 for patients aged 15 to 19; this may be explained by the high percentage of patients in that age group with advanced DMD and consequently with greater care needs.
In a more recent study, Landfeldt et al.52 analysed the economic burden of DMD in Germany, Italy, the United Kingdom, and the United States. Data were gathered using a questionnaire; 770 patients/carers completed the survey. The study estimated the direct costs of DMD in 2012 at $23920 to $54270 per patient, indirect costs associated with patients’ or carers’ loss of productivity at $18220 to $21550, and intangible costs associated with quality of life at $37980 to $46080. In summary, the total annual economic burden of DMD was estimated at $58440 to $71900 per patient. The lowest costs were found in Italy, probably due to the different organisation of the country's healthcare system.
The cost of other NMDs to society and healthcare systems has also been studied. Schepelmann et al.53 analysed health-related costs associated with MG and FSHMD in Germany using a questionnaire; a total of 41 patients with MG and 20 with FSHMD completed the survey. Total costs per patient were estimated at €26240 and €14950 per patient for FSHMD and MG, respectively. Costs were mostly due to medical insurance expenses and loss of productivity. Costs increased with disease severity and the need for a personal support worker.
Very little information is available on the economic impact of NMDs in Spain. In 2012, IMSERSO (the Spanish institute for social services and the elderly) and the Spanish Ministry of Health, Social Policy, and Equality published a study on the costs associated with rare diseases and the quality of life of affected patients.54 One of the diseases analysed in that study was DMD. A total of 57 patients completed the questionnaire. Mean age was 13.14 years (range, 3-15); 87.7% needed a carer. Mean costs amounted to €94171 (direct healthcare costs €13828, direct non-healthcare costs €79312, indirect costs €1031). Costs were mostly due to informal care (relatives or friends; €79243), outpatient care (€4211), and health and social care equipment (€3496).
Neurological care of patients with neuromuscular disordersFrom a patient care viewpoint, NMDs account for 2.8% to 18% of the reasons for consultation at neurology departments.55–57 NMDs are complex in terms of diagnosis, aetiology, and treatment, and require a multidisciplinary approach. In the case of hereditary NMDs, early diagnosis is essential to provide adequate genetic counselling. Given the high level of specialisation it requires, NMD management constitutes a subspecialty within general neurology. The Spanish Society of Neurology has a specific NMD study group, and an increasing number of journals and scientific congresses specialise in these conditions.
In this context, patients with NMDs receiving care from neurologists not specialising in NMDs may feel misinformed, anxious, or distrustful of healthcare services that cannot explain what is wrong with them. These patients may therefore seek second opinions from multiple healthcare professionals, resulting in complex and unnecessary bureaucratic and administrative processes. Clinical units specialising in ALS/NMD management include expert neurologists coordinating other subspecialties involved in the management or follow-up of these patients, such as neumology, cardiology, rehabilitation, traumatology, nutrition, genetics, physiotherapy, social services, and psychiatry. In addition to patient care, these units usually carry out significant research activities.
Neurological care for patients with amyotrophic lateral sclerosisThe diagnostic process for ALS in Spain is comparable to those used in other Western countries, including time to diagnosis. The Spanish public healthcare system offers a wide range of resources for the management of these patients. In recent years, most autonomous communities have created multidisciplinary units for ALS management. These units also conduct research on ALS and participate in international clinical trials.
Neurological care for patients with neuromuscular disordersIn most Spanish centres, patients with NMDs are cared for either in general neurology departments or in specialised NMD units; the latter usually receive patients referred by neurology departments or primary care consultations. Ideally, patients with NMDs would be followed up by these specialised units; however, this is currently not the case in Spain. Geographical differences are patent, with some patients having to travel to other autonomous communities to visit a specialised NMD unit. Furthermore, units have been created to provide transitional care from paediatrics to adult medicine. ASEM, in partnership with the Spanish Royal Board on Disability, published in 2012 the first “Map of healthcare resources for the management of neuromuscular disorders” to help patients, families, and healthcare professionals to locate specialised NMD centres in each autonomous community.58 This document provides detailed data on each centre, including information about the healthcare team, the services provided by each centre, whether these centres care for paediatric or adult patients, and whether they participate in studies or research networks.
The SEN's Neuromuscular Disorders Study Group recommends a ratio of one specialised NMD unit per million inhabitants and suggests creating one at each reference hospital.59 The specialised NMD centres already established in several autonomous communities need to be recognised as official specialised centres by the Designation Committee for Reference Centres, Services, and Units of the Spanish National Health System.
AssociationsAssociations play a key role in advising and supporting patients and promoting research into treatment. Numerous patients’ associations have been created in Spain (Appendix 1).
Amyotrophic lateral sclerosis patients’ associationsThe first national association for ALS patients in Spain (ADELA) was created in 1990. Numerous other ALS patient associations and/or delegations of ADELA have since been established in each autonomous community. Furthermore, several foundations have been created to promote research in the field of ALS, such as FUNDELA, Diógenes Foundation, Miquel Valls Foundation, and the Platform for People Affected by ALS.
Neuromuscular disorders patients’ associationsASEM has over 8000 members and has been working to raise awareness of NMDs for more than 30 years, publishing a handbook on neuromuscular diseases for families affected by ALS.60 Other important associations include the Isabel Gemio Foundation (which researches muscular dystrophies and other rare diseases), Duchenne Parent Project Spain (DMD and BMD), the Spanish myasthenia gravis patients’ association, FUNDAME (spinal muscular atrophy), and FEDER (rare diseases).
New technologies have allowed the development of numerous internet portals which streamline the exchange of information. In addition to Orphanet, which focuses on rare diseases, we should mention the role of the TREAT-NMD Neuromuscular Network, a European initiative which shares resources for patients and professionals. TREAT-NMD provides clinical management guidelines for some NMDs and information on patient registries, research projects, and clinical trials that are underway.
Conclusions- -
In Spain, NMDs account for 2.8% to 18% of reasons for consultations at neurology departments.
- -
The Survey on Disability and Dependence conducted in Spain in 2008 lists MD and ALS among the main causes of disability.
- -
ALS is the most frequent motor neuron disease in adults. Its course is progressive and invariably leads to death, typically within 5 years of onset. Incidence and prevalence of ALS in Spain are estimated at 1.4 and 5.4 cases per 100000 population, respectively. In Spain, 3 patients are diagnosed with ALS each day, and over 3000 people currently have ALS.
- -
In contrast with the relative homogeneity of ALS, other NMDs are a broad, highly heterogeneous group, which makes it difficult to address them as a whole. Many of these conditions are infrequent and are considered rare diseases, which makes it hard to clearly define the characteristics of NMDs.
- -
No data are available on the total number of patients with NMDs in Spain; however, according to ASEM, the figure may reach 60000 patients. An active patient registry created in 2010 may help expand understanding of the true impact of NMDs in Spain. At present, the Neuromuscular Disorders Database contains data on over 5000 patients with different NMDs, providing data for multicentre studies into the most prevalent disorders.
- -
ALS and most NMDs follow a chronic, progressive, debilitating course, leading to a high degree of disability and dependence. This in turn results in greater associated social and healthcare costs. The higher the degree of disability/dependence, the greater the associated costs and the lower patients’ health-related quality of life will be. The annual healthcare-related costs of ALS or DMD are estimated at $60000 per patient.
- -
NMDs are extremely complex in terms of aetiology, diagnosis, and prognosis, and require a multidisciplinary approach. Patients with NMDs should be cared for at specialised units. In Spain, this is already the case for ALS; the same is not true for other NMDs, however. In view of the evident regional differences, reference centres should receive official recognition in the Spanish national health system.
- -
Patients’ associations for ALS/NMDs play a pivotal role in our setting by providing advice and support and mediating for patients and their families.
This study has received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
ADELA: http://sites.adelaweb.com/web-adela/
Fundela: http://www.fundela.info/
Miquel Valls Foundation: http://www.elacat.org/es/
Diógenes Foundation: http://www.fundacionela.com/
Platform for People Affected by ALS: http://www.plataformaafectadosela.org/
Spanish hereditary spastic paraplegia association: http://www.aepef.org/
ASEM (federation of Spanish NMD patient associations): http://www.asem-esp.org/
Isabel Gemio Foundation: http://www.fundacionisabelgemio.com/
Duchenne Parent Project Spain: https://www.duchenne-spain.org
Spanish myasthenia gravis patient association: http://www.miasteniagravis.es/
FUNDAME (spinal muscular atrophy foundation): http://www.fundame.net/
FEDER (Spanish federation for rare diseases): http://www.enfermedades-raras.org/
Orphanet: http://www.orpha.net/consor/cgi-bin/index.php?lng=ES/
TREAT-NMD Neuromuscular Network: http://www.treat-nmd.eu/
Please cite this article as: Camacho A, Esteban J, Paradas C. Informe de la Fundación Del Cerebro sobre el impacto social de la esclerosis lateral amiotrófica y las enfermedades neuromusculares. Neurología. 2018;33:35–46.
