



El sarcoma mieloide o granulocítico es un tumor raro compuesto por células inmaduras de precursores granulocíticos, que se desarrolla fuera de la médula ósea, afectando a cualquier órgano. Se puede presentar antes, durante o después del diagnóstico de una leucemia aguda mieloide (LAM) u otra enfermedad mieloproliferativa1–4. Solo el 2-8% de los pacientes con LAM desarrollan un sarcoma mieloide, siendo extraña su aparición antes de diagnosticarse la leucemia4,5.
La World Health Organization clasifica las LAM en base al predominio celular y grado de madurez. La enfermedad extramedular se asocia a alguna de las variantes citológicas clasificadas en la French-American-British, siendo extremadamente rara en la leucemia aguda promielocítica (LAP: M3)1,2.
La LAP se caracteriza por una proliferación anormal de promielocitos y se clasifica como tipo M3 de la French-American-British. La característica citogenética de la LAP es una translocación entre los brazos largos de los cromosomas 15 y 17, y la fusión entre RARa y PML, con significado terapéutico6,7.
Solo el 10% de las LAM corresponden al tipo de LAP de buen pronóstico. Los sitios extramedulares más afectados en la LAP son la piel y el sistema nervioso central, denominándose sarcoma promielocítico1,8. El sarcoma promielocítico es extremadamente raro, soliendo presentarse como recaída de la LAP4.
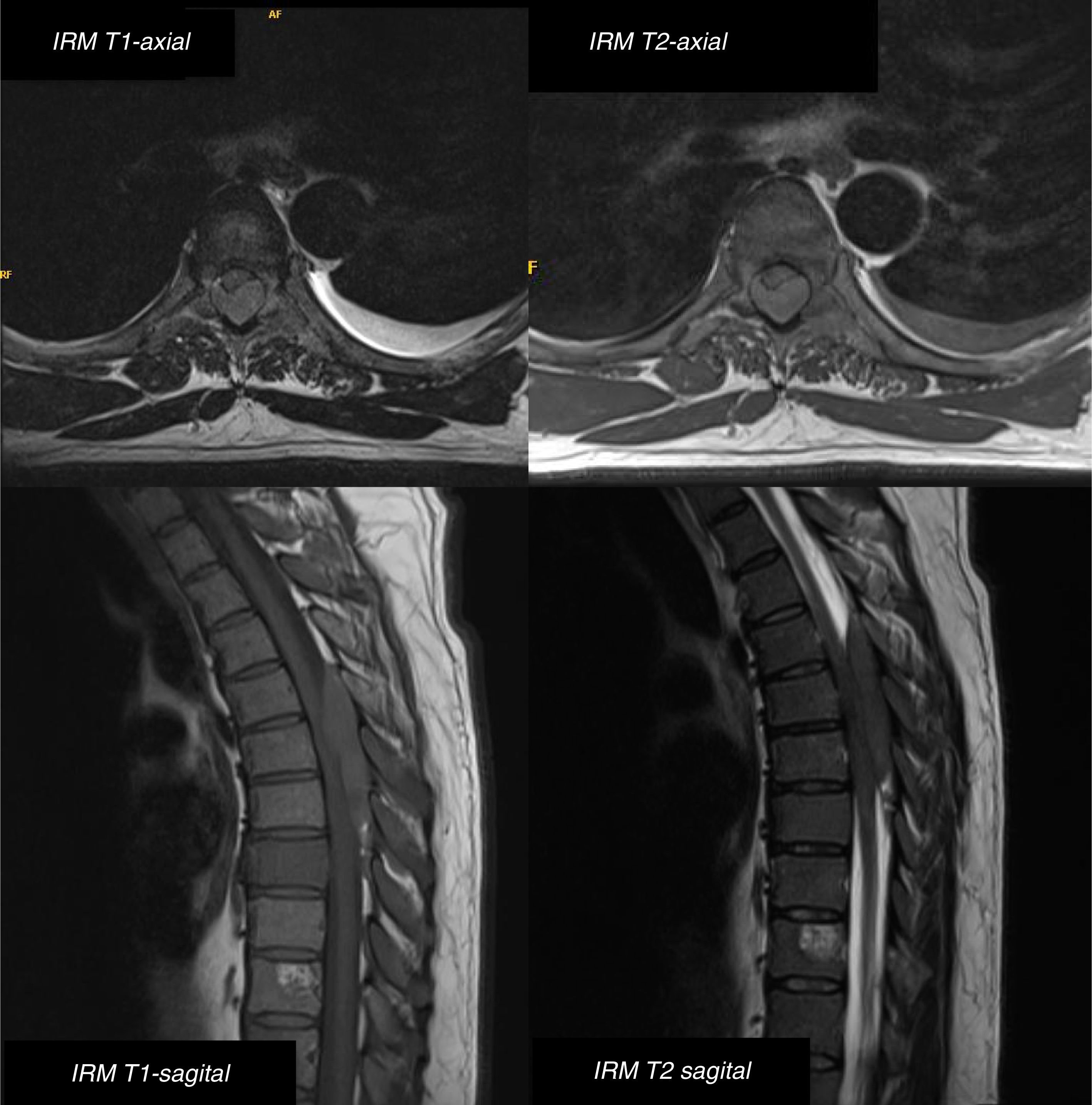
Varón de 42 años de edad con dolor óseo, derrame pleural y febrícula y de 2 meses de evolución. En menos de 24h desarrolla hematuria, paraparesia severa incapacitando la marcha, disminución de la sensibilidad en miembros inferiores y retención aguda urinaria. En la exploración destaca como patológico fuerza generalizada en miembros inferiores 2/5, reflejos osteotendinosos abolidos y reflejo cutaneoplantar indiferente bilateralmente. Asocia pérdida de sensibilidad propioceptiva y tactoalgésica con nivel T4, pérdida de tono del esfínter anal y ausencia del reflejo cremastérico; todo ello compatible con una lesión medular incompleta. La RM dorsal (fig. 1) muestra una masa epidural T5 a T8, condicionando compresión medular y múltiples lesiones óseas. El hemograma, la coagulación y bioquímica sanguínea son normales. Se realiza una laminectomía descompresiva urgente, con resección completa de la tumoración epidural. A las 42h de la cirugía el paciente desarrolla trombocitopenia, con células blásticas y promielocitos en sangre periférica. La biopsia de médula ósea es hipercelular, con infiltración de promielocitos atípicos. Mediante citometría de flujo/FISH se diagnostica de sarcoma promielocítico espinal, como presentación de una leucemia aguda promielocítica, con mofología M3 según la French-American-British con t(15;17)(q22;q21) y gen de fusión PML/RARa. El paciente es tratado acorde al Programa Español de Tratamiento en Hematología–PETHEMA LPA-AR/2011; quimioterapia de inducción (idarubicina+ATRA [all-trans retinoic acid]), quimioterapia de consolidación (idarubicin+Ara-C, citosina arabinósido+ATRA). Se administra radioterapia complementaria a nivel dorsal y se dan 2 dosis de quimioterapia intratecal triple (metotrexato+hidrocortisona+citarabina) durante las terapias de inducción y consolidación. La evolución neurológica es satisfactoria, progresiva recuperación hasta caminar con órtesis, fuerza de miembros inferiores 4/5 en caderas, 5/5 en rodillas, 4/5 en tobillos y exaltación de los reflejos osteotendinosos bilateralmente. Hipoestesia tactoalgésica inferior al nivel T10, de predominio derecho y recuperación de la función esfinteriana. Desde el punto de vista hematológico sin datos de enfermedad en LCR, ni masas paraespinales. Con aspirados de médula ósea en los que se excluye recaída, confirmándose situación de remisión completa con PML/RARa indetectable.

RM dorsal, cortes axiales y sagitales, T1 sin contraste y T2. Se aprecia masa epidural posterior isointensa desde D5 hasta D8, comprimiendo la médula en su porción posterior. Asocia múltiples lesiones óseas de la columna dorsolumbar, tanto de elementos posteriores como de cuerpos vertebrales en todos los niveles dorsales y L1.
La LAP es un subtipo de la LAM, que suele presentarse con pancitopenia y trastornos severos de la coagulación. Representa el 10-15% de todas las LAM, siendo más frecuente en Latinoamérica, España e Italia. Es la más curable de todas las LAM, fundamentalmente por la terapia con ATRA en asociación con quimioterápicos6,8,9.
Los términos sarcoma granulocítico, sarcoma mieloide o cloroma, definen aquellas raras neoplasias malignas, resultado de la proliferación extramedular de mieloblastos maduros o inmaduros en asociación a: LAM, síndromes mielodisplásicos con transformación leucemoide, leucemias mieloides crónicas con crisis blásticas, pacientes no leucémicos3.
La infiltración extramedular es una complicación muy rara de las LAM, pero es extremadamente rara en la LAP. La mayoría de los casos reportados se relacionan con una recaída de la enfermedad posterior al tratamiento, siendo el sistema nervioso central y la piel los órganos más frecuentemente afectados. Los factores asociados a la recurrencia extramedular son edad menor a 45 años, recuento elevado de células blancas, la isoforma bcr3 del gen de fusión PML/RARa y el tratamiento con ATRA. Es extraordinaria la presentación con enfermedad extramedular, sin datos de leucemia5,6,9.
La terapia con ATRA en pacientes con LAM facilita la aparición del sarcoma granulocítico, debido a un aumento en la expresión de moléculas de adhesión en los promielocitos leucemoides y sus ligandos en las células endoteliales. También se cree que podría facilitar el paso de promielocitos malignos a través de la barrera hematoencefálica, predisponiendo a recidivas en sitios poco frecuentes6.
El diagnóstico del sarcoma granulocítico es fácil cuando este es sincrónico a una LAM o como recaída de una LAM tratada. Sin embargo el diagnóstico es complicado cuando el sarcoma granulocítico precede a la instauración de la LAM. Más del 75% de estos sarcomas son erróneamente diagnosticados como otro tipo de neoplasias, principalmente linfomas malignos. Se recomienda el estudio con microscopia electrónica, tinciones para la diferenciación mieloide en conjunto con naftol-AS-D cloroacetato, mieloperoxidasa, tinción de inmunoperoxidasa para lisozima y CD34 junto con otros marcadores para células B y T, particularmente CD79a y CD32,3,5,6,10.
Los pacientes con sarcoma granulocítico aleucémico desarrollarán eventualmente una leucemia aguda. Se estima que entre el 66-88% de estos pacientes presentará una leucemia mieloide aguda 9-11 meses después del diagnóstico3.
El tratamiento del sarcoma granulocítico en ausencia de leucemia deberá incluir altas dosis de quimioterápicos específicos para LAM, con o sin radioterapia3,5. Cuando se dejan sin tratar, la mayoría de los sarcomas granulocíticos primarios evolucionarán a una leucemia. Aquellos tratamientos consistentes en procedimientos locales, incluyendo la resección tumoral y radioterapia sin quimioterapia, tendrán un mayor riesgo de enfermedad sistémica5.
El tratamiento estándar consiste en la combinación de ATRA con quimioterapia del grupo de las antraciclinas. Las tasas de remisión completa de la enfermedad son del 85-95% con una supervivencia a los 5 años del 65-70%, tanto en niños como en adultos5. El programa español PETHEMA ha demostrado tasas de remisión significativamente mayores en comparación con otros grupos, asociando menor número de muertes durante la terapia de inducción y consolidación. Asimismo la supervivencia libre de enfermedad, tras alcanzar la remisión completa, es comparable e incluso superior a otros protocolos establecidos11.
Existen cientos de casos reportados de sarcomas granulocíticos asociados a diferentes subtipos de LAM, pero solo hay 8 casos de sarcoma granulocítico con LAP, 3 de los cuales se presentaron en pacientes no leucémicos2.
En conclusión, el diagnóstico temprano y el tratamiento adecuado son fundamentales para obtener resultados satisfactorios.

