



La siderosis meníngea (SM) es una entidad rara debida al depósito de hemosiderina secundario a sangrados recurrentes en la capa subpial de las meninges. Estos depósitos producen de manera progresiva proliferación glial, fibrosis y finalmente la lesión de las neuronas de la superficie cerebral. El síndrome periódico asociado al receptor del TNF (TRAPS) es una enfermedad autoinflamatoria con herencia dominante caracterizada por episodios prolongados de fiebre, mialgias y exantema migratorios, serositis asépticas y elevación de reactantes de fase aguda.
Paciente varón de 46 años con retraso mental moderado, no filiado, con un grado de disminución psíquica del 65%. Entre sus antecedentes familiares destaca una amiloidosis secundaria en su padre, portador de la variante heterocigota p.Arg92Gln del gen TNFRSF1A. Entre sus antecedentes personales destacan crisis comiciales a los 15 años, sin necesidad de tratamiento actual, y presbiacusia bilateral (OD 64%; OI 37%). Consulta por inestabilidad progresiva, de 2 años de evolución, envejecimiento prematuro, abasia grave, sin lentitud ni dificultades en el giro, desorientación temporoespacial y con cambios de humor frecuentes. Presentó un test de Romberg positivo, no objetivándose disartria, alteraciones de la deglución, diplopía, nistagmo, focalidad motora de vías largas ni dismetrías en extremidades. Todos los estudios hematológicos y bioquímicos realizados, incluyendo proteinograma y niveles plasmáticos de reactantes de fase aguda, fueron normales o negativos. Asimismo, el análisis del gen TTR no demostró variantes patogénicas.
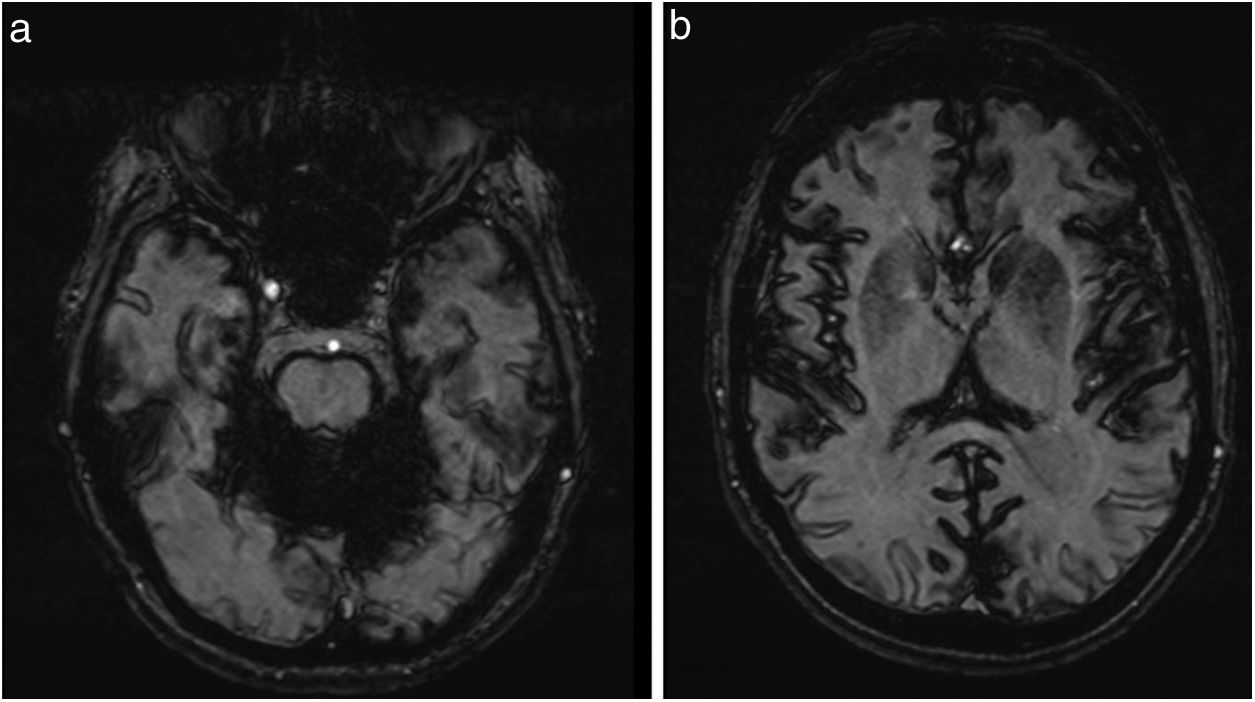
La ecografía abdominal y la colonoscopia fueron normales, mientras la gastroscopia evidenció una hernia de hiato. Las biopsias de duodeno y recto no presentaban depósitos amiloideos. La RMN cerebral demostró atrofia cerebelosa, especialmente de vermis, y depósitos difusos de hemosiderina en fosa posterior (fig. 1a) y, en menor grado, en surcos cerebrales (fig. 1b), con marcada susceptibilidad magnética en las secuencias de gradiente. El electromiograma fue normal.

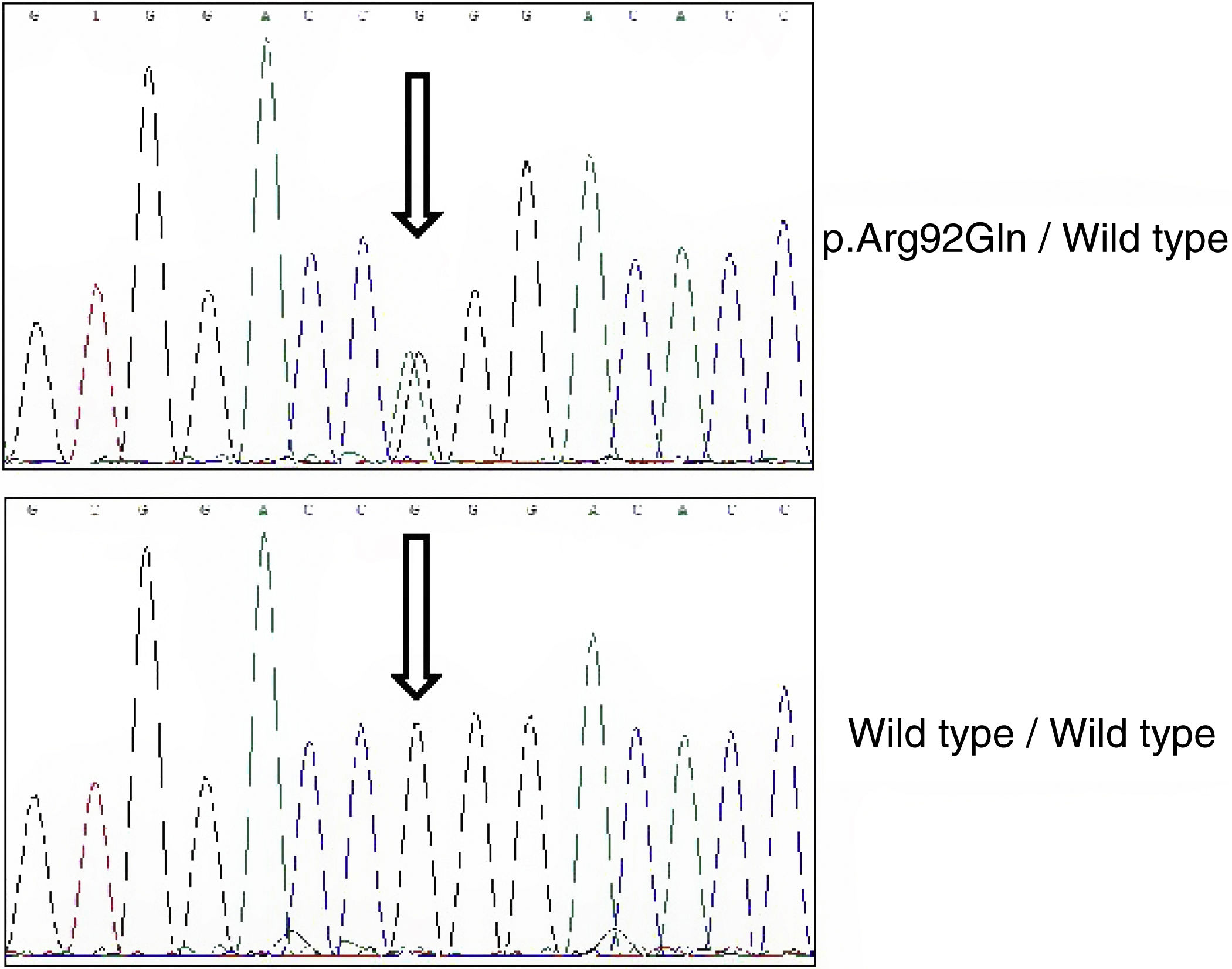
En base a sus antecedentes familiares se realizó el estudio genético mediante un panel de genes responsables de enfermedades autoinflamatorias que incluía los genes MEFV, TNFRSF1A, MVK, NLRP3, NOD2, PSTPIP1 y CECR1. La librería se generó mediante amplificación de todos los exones y las regiones intrónicas adyacentes. La secuenciación se realizó en la plataforma NextSeq (Illumina), y entre los criterios de análisis de las secuencias obtenidas se incluía una cobertura mínima 50x. Asimismo, todas las variantes clasificadas como patogénicas, probablemente patogénicas o de significado incierto fueron confirmadas mediantes secuenciación tipo Sanger. El estudio reveló un genotipo heterocigoto sencillo para la variante p.Arg92Gln (fig. 2).

La SM es una entidad clínica poco frecuente. Antes del advenimiento de la RMN, su diagnóstico se realizaba incidentalmente, bien en intervenciones quirúrgicas, bien en necropsias. Actualmente, las imágenes de RMN son patognomónicas, permitiendo obtener su diagnóstico incluso en fases tempranas. El hallazgo más característico son las hipointensidades lineales en secuencias T2 a lo largo de las superficies leptomeníngeas vermianas y cerebelosas, y de forma menos extensa las superficies leptomeníngeas cerebrales.
En un 50% de los casos, su causa es desconocida1, mientras que en otros se ha relacionado con hemorragia cerebral asociada a tumores, a la angiopatía amilodea2,3 y a malformaciones vasculares4,5. En el caso aquí presentado no se demostró ninguna de estas causas, incluyendo la amiloidosis leptomeníngea, una forma de presentación clínica rara entre las amiloidosis hereditarias por mutaciones en el gen TTR6. En el paciente aquí descrito se ha detectado una variante en el gen TNFRSF1A. Mutaciones en este gen originan el TRAPS, una enfermedad autoinflamatoria con herencia dominante caracterizada por episodios prolongados de fiebre, mialgias y exantema migratorios, serositis asépticas y elevación de reactantes de fase aguda7. Ocasionalmente, durante la edad adulta, algunos pacientes ven complicado el curso de su enfermedad con el desarrollo de amiloidosis de tipo AA8.
Lo intrigante del presente caso es la coexistencia de la SM con la variante p.Arg92Gln del gen TNFRSF1A. Por un lado, en el paciente aquí descrito no se han objetivado episodios febriles recurrentes ni tampoco hay evidencias de depósito amiloideo de tipo AA. La edad de inicio del TRAPS es mayormente pediátrica, aunque en algunos portadores de la variante p.Arg92Gln, la sintomatología puede aparecer en la edad adulta9. Finalmente, los pacientes portadores de esta variante son un grupo clínicamente heterogéneo, desde individuos asintomáticos/presintomáticos hasta pacientes que van a precisar tratamientos biológicos antiinflamatorios.
En un trabajo previo se demostraron cambios microangiopáticos a nivel cerebral en pacientes afectos de TRAPS10. A pesar de ello, no hay evidencias claras de las causas que pueden relacionadas la aparición de la SM con este síndrome. A modo de hipótesis, se podría pensar en una alteración de la permeabilidad de la barrera hematoencefálica. En el TRAPS se observa un aumento marcado de múltiples citocinas inflamatorias circulantes. Entre estas destaca la IL-1beta, hasta el punto que los tratamientos bloqueantes de esta citocina son en la actualidad los más efectivos en su tratamiento11. A nivel de la barrera hematoencefálica, el aumento de IL-1 produce un incremento de la MCP-1, que puede provocar una alteración de las uniones estrechas de la barrera hematoencefálica, conllevar un aumento de su permeabilidad12, que alteraría el transporte de transferrina a través de ella y como consecuencia aumentaría el depósito de de ferritina en el tejido cerebral.
En resumen, presentamos el primer caso descrito de un paciente en el que concurren SM y la variante p.Arg92Gln del gen TNFRSF1A. Debido a la posible existencia de una relación entre ambas entidades, creemos conveniente incluir esta enfermedad inflamatoria monogénica en el diagnóstico diferencial de la SM.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.

