



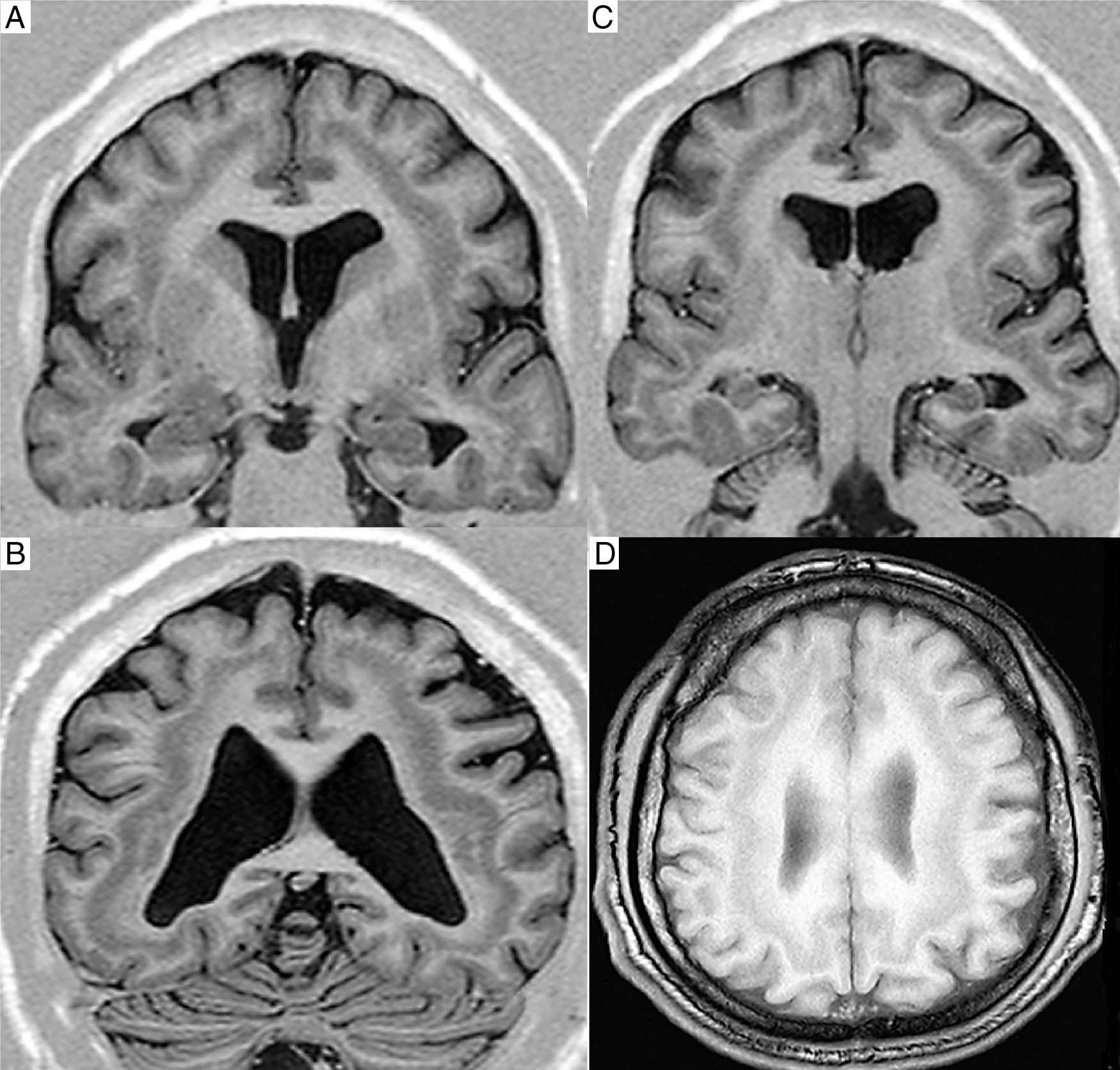
La heterotopía en banda subcortical (HBS), también conocida como síndrome de doble corteza (SDC) es un inusual trastorno de la migración cortical, caracterizado por una bien delimitada capa de sustancia gris, en la sustancia blanca, entre la corteza cerebral y los ventrículos laterales. Posterior a la introducción de la imagen por resonancia magnética (IRM) en la práctica clínica fue posible identificar y caracterizar esta entidad1. La mayoría de los casos descritos pertenecen al sexo femenino, con escasos reportes en varones, los que suelen presentar generalmente lisencefalia asociada, con afectación neurológica más severa2.
Paciente masculino de 26 años que es referido a nuestro centro por sospecha de epilepsia refractaria. Hijo de matrimonio no consanguíneo, sin antecedentes prenatales relevantes, producto de parto a término sin complicaciones, alcanzando hitos del neurodesarrollo acordes durante su primer año de vida. Sin antecedentes familiares de epilepsia, ni de discapacidad intelectual. Durante el inicio de la vida escolar tuvo un bajo rendimiento en sus calificaciones, con dificultades en el aprendizaje, por lo que en evaluación psicológica se diagnosticó retraso mental leve-moderado. A los 6 años de edad comienza con crisis convulsivas, episodios de desconexión que en ocasiones evolucionaban a pérdida de la conciencia con movimientos de todo el cuerpo. Durante los últimos 20 años, las crisis incrementaron en frecuencia, y aparecieron crisis mioclónicas, tónico clónicas generalizados y atónicas. Al examen clínico destacaba rasgos dismórficos como hipertelorismo y puente nasal ancho. Utilizó tratamiento con múltiples antiepilépticos, fenitoína, carbamazepina, clonazepam, clobazam, ácido valproico y lamotrigina, en monoterapia, biterapia y politerapia, sin alcanzar un control adecuado (figs. 1 y 2). Se inició politerapia racional con lamotrigina 300mg/día, ácido valproico 2,5g/día y levetiracetam 3g/día teniendo un control aceptable con solo 2 crisis parciales en los últimos 6 meses.
 (A-C), cortes coronales e imagen T1 corte axial (D) donde se evidencia banda de sustancia gris en la sustancia blanca subcortical, sin lisencefalia o agiria-paquigiria.")

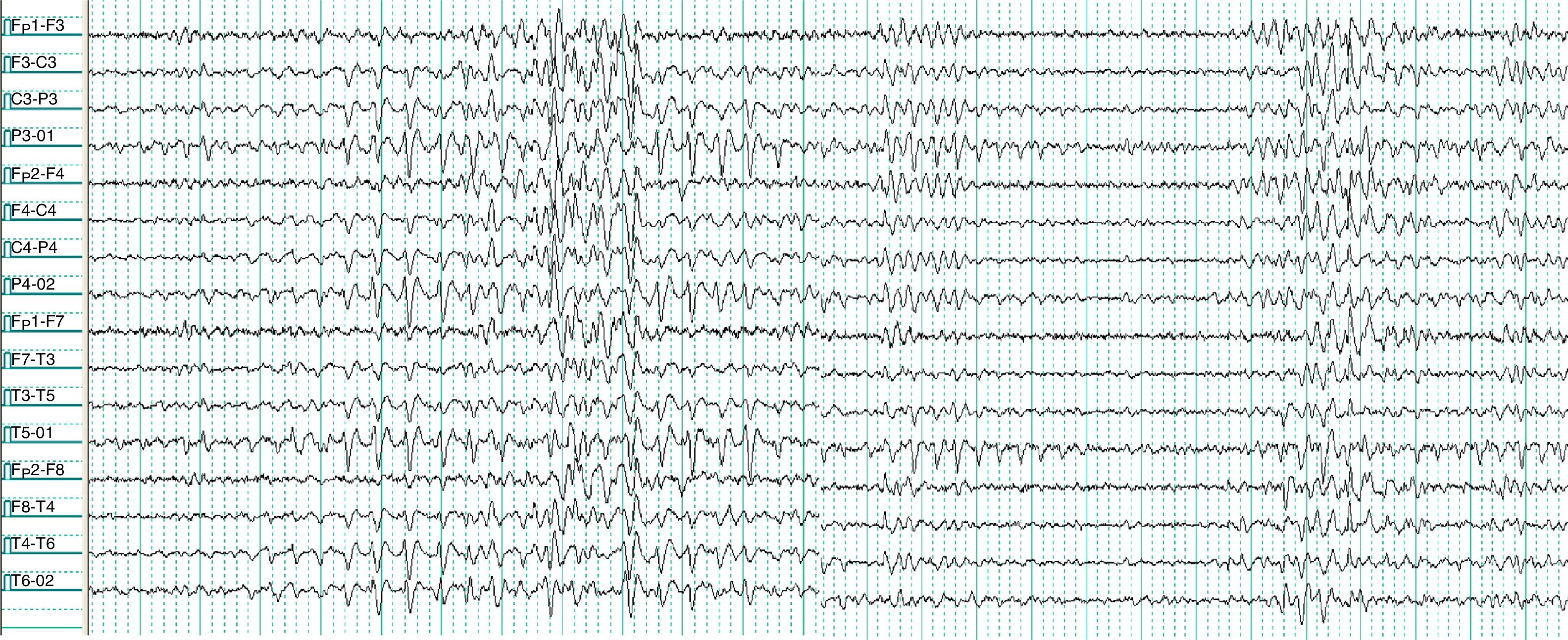
Electroencefalograma vigilia, sistema internacional 10-20, montaje bipolar longitudinal. Donde apreciamos etapas del registro, inicialmente actividad paroxística que inicia en región occipital izquierda, después en regiones frontales manteniéndose la actividad en regiones occipitales de forma independiente, predominantemente del hemisferio izquierdo durante todo el registro.
El SDC es un trastorno poco frecuente de la migración cortical asociado a mutaciones de los cromosomas 17p13.3 (LIS1) y ligada al Xq22.3-q23 (DCX o XLIS). Las mutaciones del gen de doblecortina (DCX) representa el 85% de los casos esporádicos del sexo femenino y el 25% del sexo masculino1,3. Este gen desempeña un papel importante en la codificación de las proteínas de los microtúbulos que intervienen en la migración neuronal durante el desarrollo cerebral, por lo que mutaciones del mismo provoca un severo daño de la estructura de la corteza cerebral4. Las características clínicas que nos permiten sospechar del SDC son el inicio de crisis epilépticas en la primera década de vida, que evolucionan a la refractariedad paulatina, con múltiples tipos de crisis y discapacidad intelectual5. La HBS tiene mayor frecuencia en el sexo femenino, cuando se presenta en pacientes masculinos suelen tener asociado lisencefalia o agiria-paquigiria en regiones frontales, predominantemente2,6. Alteraciones que no se evidencian en este paciente. Escasos son los reportes de pacientes masculinos con SDC sin lisencefalia o agiria-paquigiria. El hallazgo de mosaicismo para la mutación del gen DCX, es una posible explicación de menor severidad de las manifestaciones clínicas en varones6. Poca es la evidencia existente sobre el tratamiento de esta entidad cuando la politerapia racional falla. El tratamiento quirúrgico, pese a tratarse de una epilepsia con actividad focal ha mostrado resultados desalentadores7. Recientemente se ha descrito en 2 pacientes que la estimulación bilateral de los núcleos anteriores del tálamo puede contribuir a mejorar el control de las crisis epilépticas8.

