



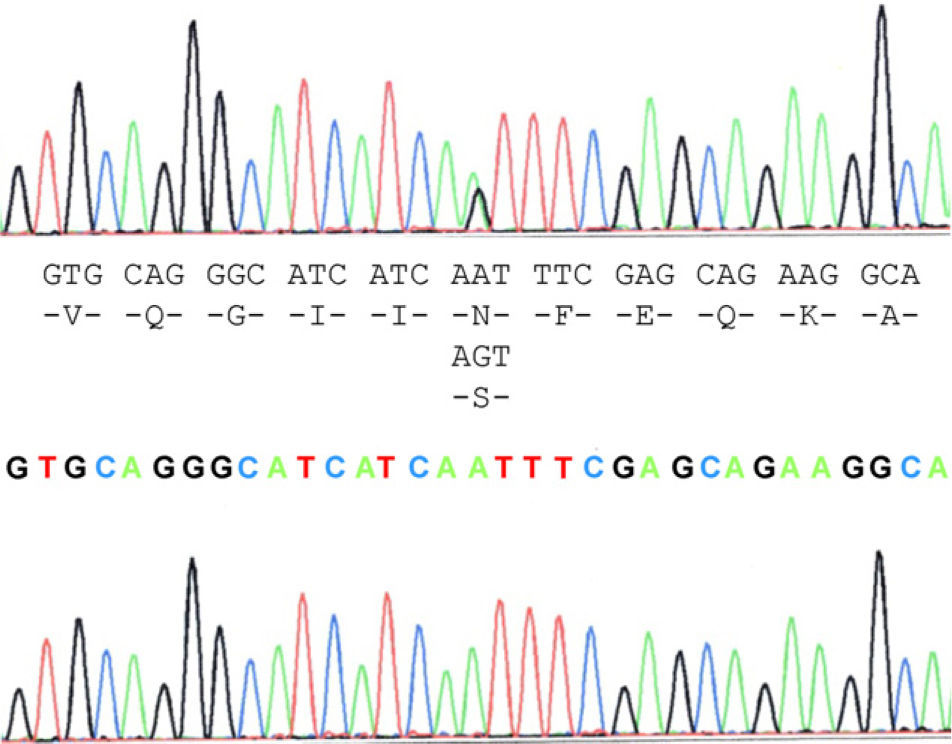
N19S mutation is produced by substitution in the 139 position of SOD1 and was described by Mayeux in a patient with amyotrophic lateral sclerosis (ALS). He suggested that it did not have a causal effect as it was found in asymptomatic and sporadic cases. Other authors in later articles did not agree.
Material and methodsWe describe a family with 4 members with ALS patients and attempt to find the carrier of the N19S mutation of the propositus. Molecular studies were performed on 15 members of the family of a different order.
ResultsThe ALS cases were found in the maternal line of the propositus. The presence of the mutation was detected in 3 people; the other two were asymptomatic. One of patients with ALS in the family, who died previously, did not have the mutation. Two of the sons of this case and another of the other case did not show it. On the other hand, N19S mutation was only present in paternal branch of the propositus, where there were no cases.
ConclusionThe described family supports the hypothesis by Mayeux and against that mutation N19S has pathological consequences, since mutation is only in the family line where there are no cases with ALS. In consequence, although the described case is included as a familiar form, it cannot be attributed to the mutation, and its relationship with N19S should be considered as casual.
La N19S es una mutación por la sustitución en la posición 139 de la SOD1 y fue descrita por Mayeux et al, donde los autores sugirieron que no tenía un efecto causal al hallarse casos asintomáticos y esporádicos pero autores posteriores han sugerido lo contrario.
Material y métodosSe describe una familia con 4 pacientes con ELA en los que en el caso propósito es portador de la mutación N19S. Se realiza un estudio molecular en 15 personas de la familia de diferente orden.
ResultadosLa enfermedad se halla en la línea materna del primer caso. Se detecta la presencia de la mutación en tres personas, el primer y dos asintomáticos. Uno de los pacientes afectos de ELA de la familia que murió previamente, no presentaba la mutación. Dos de los hijos del tercer caso y otro del cuarto caso tampoco la mostraron. Contrariamente, la mutación está presente en la rama paterna del primer caso, que es asintomática.
ConclusiónLa familia descrita apoya la hipótesis de Mayeux et al. y va en contra que la mutación N19S tenga connotaciones patológicas, ya que la mutación se encuentra en la línea familiar no afecta de la enfermedad y no está en la línea con enfermos de ELA. En consecuencia, aunque el caso descrito es una forma familiar no puede ser atribuido a la mutación y su relación debe ser considerada como casual.
Up to 10% of cases of amyotrophic lateral sclerosis (ALS) are familial1 and, of these, approximately 25% are associated with a mutation in the superoxide dismutase-1 gene (SOD1),2 of which over 100 different types have been described.3,4 The production of an abnormal SOD1 protein by these mutations could trigger the mechanisms of apoptosis and neuronal lesion.5 In addition, transgenic experimental models of familial ALS were obtained through some of them,6,7 thus leading to therapeutic lines being postulated.8 However, there is dispute that they may be applicable to sporadic patients.9,10 Although most described familial forms associated with SOD1 mutations are dominant,11 some with a recessive inheritance pattern have been observed.12 Consequently, some patients have been described who could be sporadic13–16 or carrying heterozygous forms with incomplete penetrance.17,18 However, it could also be suggested that they may be asymptomatic or have no pathological significance. The N19S mutation is caused by a substitution of ASN by SER at position 139, in exon 1; its first description was by Mayeux et al.19 in 2003, who considered that it might be asymptomatic. In the current work, we report a case in an ALS family who presented the N19S mutation.
Materials and methodsCase studyThe patient studied was a female, 52 years old at the time of diagnosis, without relevant medical history, who began to suffer progressive weakness and amyotrophy in the distal segments of her right upper limb. In addition to paresis and atrophy of distal muscles, the initial neurological examination found fasciculations and hyperreflexia in the affected limb. She presented no sensory deficits or other associated neurological signs. The electromyogram (EMG) found fasciculations and spontaneous denervation activity, as well as loss of motor units in the voluntary pathways of maximum effort in several muscles of the right upper limb. Apart from that, the neurophysiological study did not detect conduction blocks and nerve conduction velocities were within normal ranges. A magnetic resonance imaging (MRI) of the brain and cervical spine was conducted, finding no structural damage. Other complementary tests were also normal, including haemogram and blood chemistry profile, immunoelectrophoresis, thyroid hormones, vitamin B12 levels, serology for Borrelia, syphilis and HIV, and antinuclear, anti-ganglioside and antineuronal antibodies, as well as a cytobiochemical study of cerebrospinal fluid. Riluzole was prescribed from the early stages of clinical symptoms, at a daily dose of 100mg. Motor symptoms and signs progressed and gradually spread to proximal muscles, although this progression was slow. Finally, 3 years after onset, clinical manifestations spread to the remaining limbs. At that time, neurological examination revealed the presence of bilateral motor signs, with weakness, fasciculations and muscle atrophy in both upper limbs, fasciculations in both quadriceps femoris and universal hyperreflexia. In these stages, the EMG revealed a chronic neurogenic pattern in the muscles of all limbs. Bulbar muscles were not yet affected.
Family studyThe patient had a family history of disease through her mother. One uncle and 2 of his children had suffered ALS with the same clinical criteria. The paternal line of this family consisted of 6 first-order individuals and 18 second-order. The maternal line was composed of 9 individuals of the first order and 19 of the second order. To this we must add the case in question and her 5 siblings. Of the 58 family members between the first and second orders, we studied 12 people, plus 3 more of the third order.
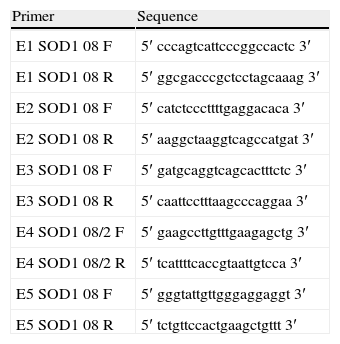
Molecular analysisBlood samples from the patient, relatives and controls were obtained with informed consent. DNA extraction was performed by PCR using standard procedures to expand 5 exons of the SOD1 gene (Table 1). The detection of variations in the sequences was performed using sequencing. The PCR products were purified with the GFX PCR DNA purification kit (Amersham Pharmacia Biotech, Piscataway, NJ). Direct sequencing of PCR products was performed with the same set of primers (anteverted and reverse) in an ABIPrism 310 DNA sequencer using the Cycle Dye Terminator DNA sequencing kit from Applied Biosystems (Perkin Elmer-Applied Biosystems, Foster City, CA). The analyses were performed using sequence analysis software with the GenBank sequence reference L44135–L44139.
List of primers used to obtain the 5 PCR products corresponding to the 5 SOD1 exons and their flanking 5′ and 3′ sequences.
| Primer | Sequence |
| E1 SOD1 08 F | 5′ cccagtcattcccggccactc 3′ |
| E1 SOD1 08 R | 5′ ggcgacccgctcctagcaaag 3′ |
| E2 SOD1 08 F | 5′ catctcccttttgaggacaca 3′ |
| E2 SOD1 08 R | 5′ aaggctaaggtcagccatgat 3′ |
| E3 SOD1 08 F | 5′ gatgcaggtcagcactttctc 3′ |
| E3 SOD1 08 R | 5′ caattcctttaagcccaggaa 3′ |
| E4 SOD1 08/2 F | 5′ gaagccttgtttgaagagctg 3′ |
| E4 SOD1 08/2 R | 5′ tcattttcaccgtaattgtcca 3′ |
| E5 SOD1 08 F | 5′ gggtattgttgggaggaggt 3′ |
| E5 SOD1 08 R | 5′ tctgttccactgaagctgttt 3′ |
The case study patient who met the criteria for ALS20 was found to be carrying the N19S mutation (Fig. 1). Of the family members studied, 3 of them (the patient and 2 asymptomatic people, the father aged 87 years and a brother aged 48 years) were also carriers of the mutation. Apart from the case study patient, the biological material from an ALS patient in the family (case 3), who had died and whose report had been filed previously, revealed that this patient did not carry the mutation. Two of the 5 children of case 3 did not show the presence of the mutation, and neither did 1 of the 3 children of case 4. Contrary to expectations, the study showed that the mutation was present in the paternal side of the case patient, that is, that branch that did not have cases of ALS. This suggests that the familial form of ALS is not subject to the mutation, except for the case patient, who obtained it from the maternal line, whereas the mutation was present in the paternal line that had no patients with ALS. This suggested that the disease may not be related with the mutation and its presence would be coincidental.
Discussion
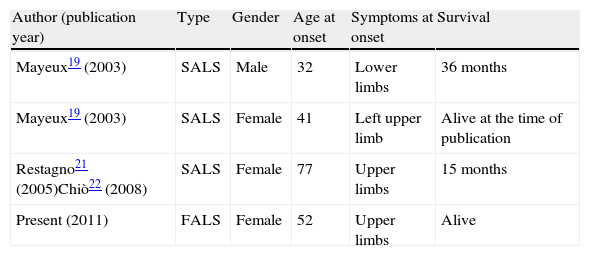
The N19S mutation was described by Mayeux et al.19 and some more cases were subsequently reported21,22 but not in familial forms (Table 2). The original study described 7 family members who presented the mutation with no symptoms, out of 15 subjects studied in 3 generations (between 30 and 80 years old). This led the authors to suggest that carrying the mutation was not the cause of disease, since they also found a subject carrying the mutation amongst 180 controls.
Cases of ALS associated to the N19S mutation described in the literature.
| Author (publication year) | Type | Gender | Age at onset | Symptoms at onset | Survival |
| Mayeux19 (2003) | SALS | Male | 32 | Lower limbs | 36 months |
| Mayeux19 (2003) | SALS | Female | 41 | Left upper limb | Alive at the time of publication |
| Restagno21 (2005)Chiò22 (2008) | SALS | Female | 77 | Upper limbs | 15 months |
| Present (2011) | FALS | Female | 52 | Upper limbs | Alive |
ALS: amyotrophic lateral sclerosis; FALS: familial ALS; SALS: sporadic ALS.
The role of SOD1 mutations in ALS is currently under discussion.10,23 The possibility has been noted that the same mutation may have different penetrance and that the most characteristic example is the mutation SOD1-D90A.24,25 This mutation has 2 phenotypic forms; 1 homozygous form (described in Torne Valley, between Sweden and Finland) and 1 heterozygous form (found in other European countries).26 Homozygous forms are benign, characterised by pyramidalism and long survival, whereas heterozygous forms are much more aggressive and similar to the dominant familial forms, like carriers of other SOD1 mutations.27 In this sense, the SOD1-N19S mutation can also present different phenotypes. Another possibility that might explain a different phenotype with the same mutation could be associated with a potential reduction of the mutation load. This has been demonstrated in transgenic mice, in which, as generations pass, the disease is modified and there is even an increase in survival.28
In the original publication describing the N19S mutation, Mayeux et al.19 suggested that the association was not causal, given that the AA substitution occurred in a location with little relevance in the molecule. These authors considered that N19 was not conserved in different species and that, therefore, it should not have a significant role in the function of SOD1 and, consequently, its replacement could only generate minor modifications. However, Obata et al.29 conducted a pilot study with the mutation in which they transfected cDNA-N19S-SOD1 to NSC34 cells, which are hybrid cells of embryonic spinal cord motor neurons from mice and cells from mouse neuroblastoma. They found that overexpression of N19S-SOD1 induced neuronal death in these cells, that intracellular aggregates occurred in a low proportion (higher than in controls, but lower than in the most common SOD1 mutations) and that they showed oxidative stress that increased polymerisation through oxidative treatment, although to a lesser degree than other mutations, with accelerated polyubiquitination, in a manner similar to other SOD1 mutations. From this, they concluded that the N19S mutation caused motor neuron death and generated a mutated protein, which suggested that the disease probably occurred in combination with other risk factors.
On the other hand, our studied family supports the hypothesis of Mayeux et al.,19 which is contrary to the idea that the presence of the mutation has pathological connotations. The mutation was present in the family line not affected by the disease and was not present in the line with ALS patients. This situation is similar to that described by Felbecker et al.30 for the D90A and E100K mutations, after finding ALS patients with and without mutations within the same family. Consequently, although our case is a familial form, it cannot be attributed to the mutation and its relationship should be considered as coincidental, thus confirming the idea that the association with an SOD gene mutation may be asymptomatic.31
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Vela A, et al. Mutación SOD1-N19S en una familia de esclerosis lateral amiotrófica. Neurología. 2011;27:11–15.
