



Osmotic demyelination syndrome is a rare, severe neurological disease that includes extrapontine and central pontine myelinolysis (CPM), with the latter being the most frequent form. The condition was first described in 1959 by Adams et al.1 It is typically observed in alcoholic patients and patients with liver transplant or in a hyperosmolar state (particularly after rapid correction of chronic hyponatraemia). However, the syndrome has been also described in situations of severe hyperosmolar state not associated with hyponatraemia, including diabetes mellitus, severe hypophosphataemia, hypokalaemia, kidney failure, haemodialysis, hyperemesis gravidarum, anorexia nervosa, Wilson disease, severe burns, systemic lupus erythematosus, acute intermittent porphyria, cytomegalovirus hepatitis, Epstein-Barr virus–associated haemophagocytic lymphohistiocytosis, anaphylactic shock, and heatstroke.2,3 Apparently, any sudden alteration in the hyperosmolar state may trigger onset of this syndrome.
We present the case of a 52-year-old man with personal history of type 2 diabetes mellitus with associated microvascular complications; poor metabolic control (last recorded value of glycosylated haemoglobin of 11.7% 4 months earlier); dyslipidaemia; history of intravenous drug use (with no other substance abuse); stage C3 HIV infection with good immunovirological condition; and decompensated hepatic cirrhosis due to HCV genotype 3 infection, which resolved with sofosbuvir and daclatasvir but presented persistent severe complications in recent months (episodes of hepatic encephalopathy, bleeding gastric varices, and fluid retention) as a consequence of a very advanced cirrhosis. The patient was awaiting assessment to be placed on the waiting list for liver transplantation.
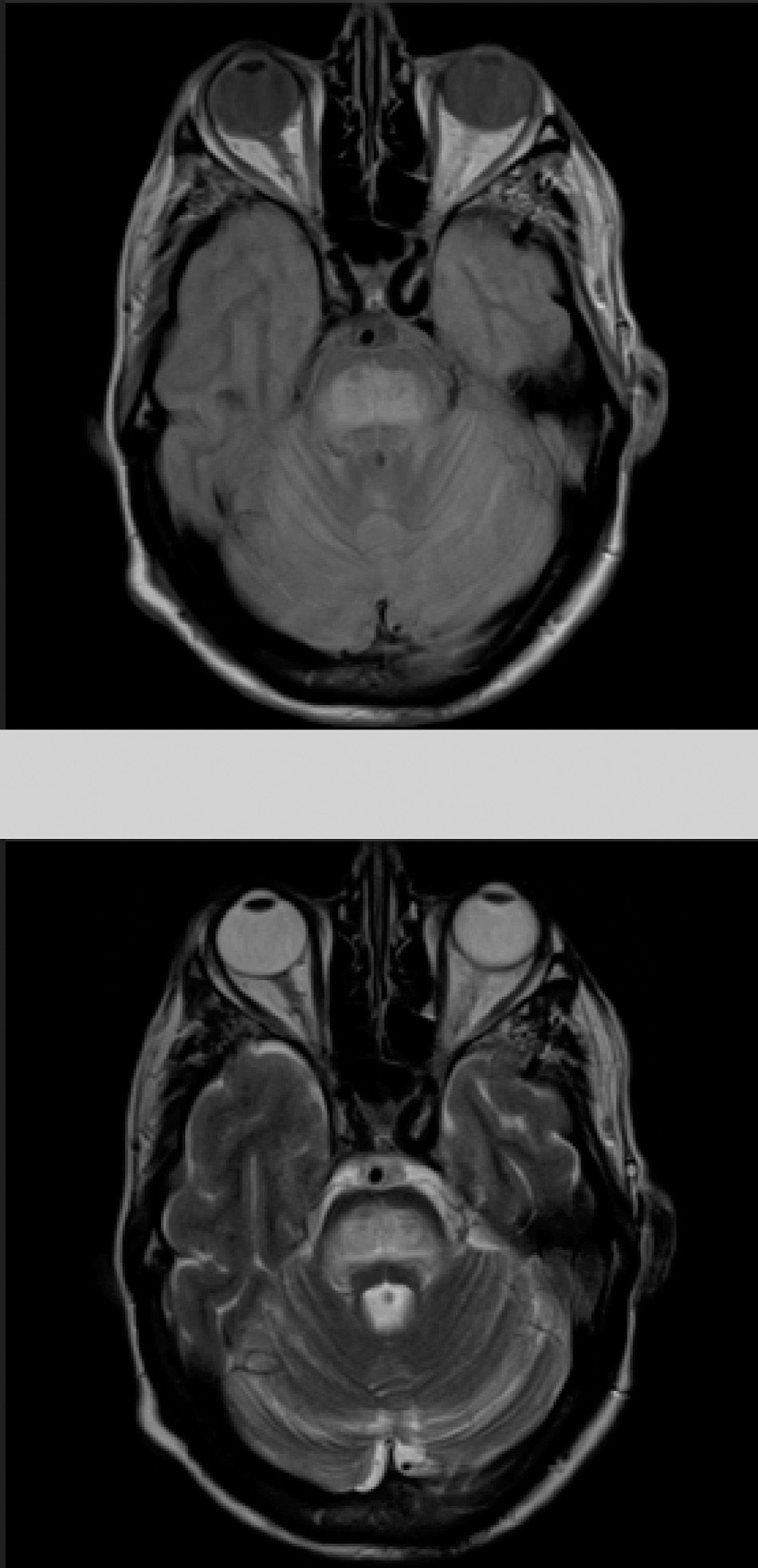
The patient also presented a 2-month history of dysarthria, dysphagia, and gait instability. The physical examination revealed moderate dysarthria, limited upgaze, and marked truncal ataxia. A laboratory analysis performed at admission showed glucose concentration of 406mg/dL, creatinine of 1.37mg/dL, Na 133mmol/L, K 4.62mmol/L, and no other relevant findings. A brain computed tomography study revealed no abnormalities; the cerebrospinal fluid analysis performed within 24 hours of admission (biochemical [including oligoclonal bands], cytological, and microbiological studies [polymerase chain reaction] testing for herpes viruses 1 and 2, Listeria antigens, meningococcus, pneumococcus, Haemophilus, enterovirus, Epstein-Barr virus, varicella zoster virus, cytomegalovirus, fungal infection, HIV viral load, and mycobacteria) yielded no pathological results. A brain magnetic resonance imaging (MRI) study (Fig. 1) performed at 48 hours of admission showed a hyperintense lesion to the pons on proton-density/T2-weighted and FLAIR sequences, with discrete diffusion restriction; all these findings are compatible with CPM.

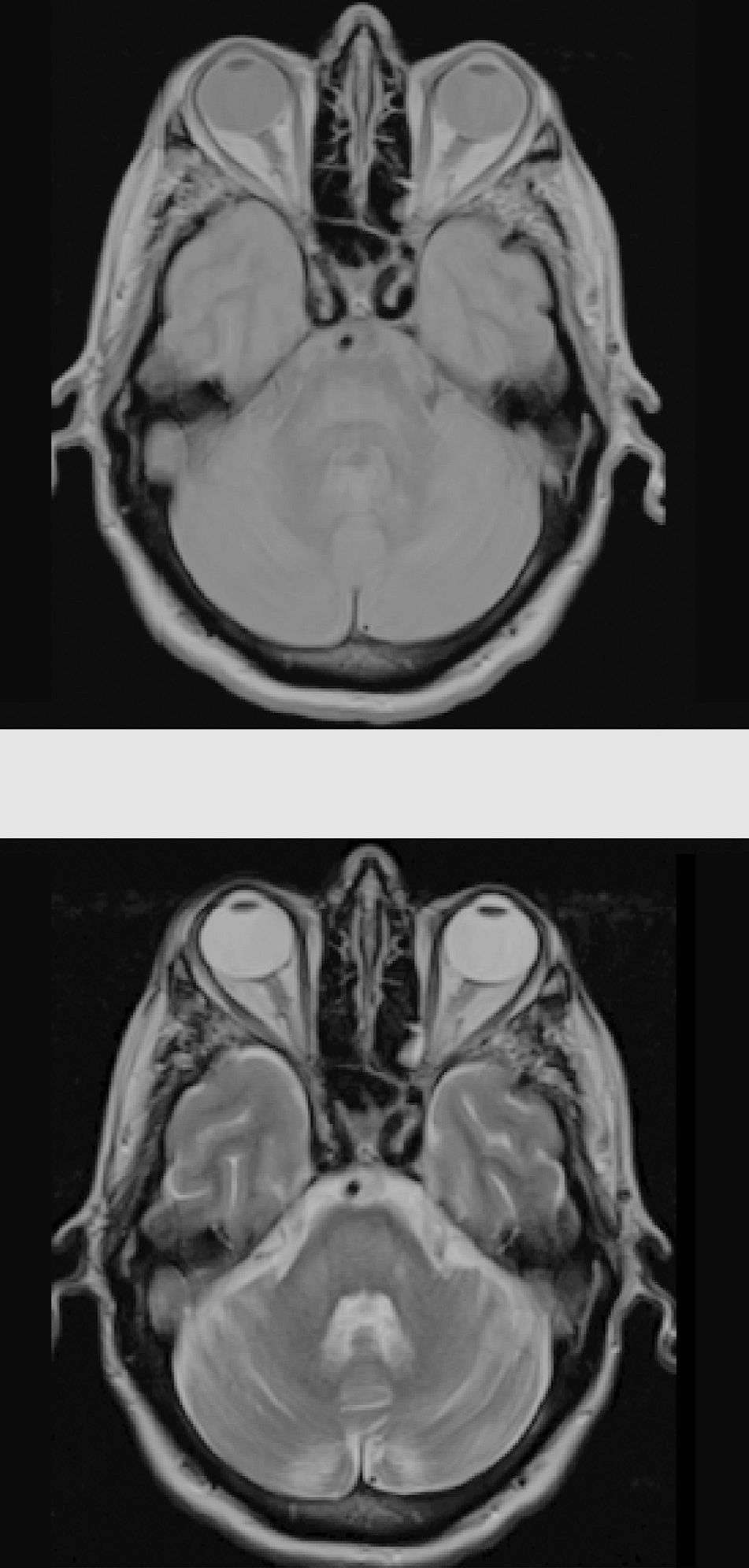
Considering the patient's history, we concluded that he had presented resolved hyponatraemia that was not detected in previous analyses, with only high glycaemic values (around 500-600mg/dL) persisting; this would cause a hyperosmolar state, triggering CPM. The remaining analytical parameters were normal (biochemistry, ions, autoimmunity, tumour markers, antineuronal antibodies, and toxicology). During hospitalisation, glycaemic levels were well controlled, with progressive neurological improvement; at discharge, the patient presented mild dysarthria only. A 5-month follow-up examination showed complete resolution of neurological symptoms, and a brain MRI scan displayed clear lesion improvement (Fig. 2).

The pathophysiological mechanisms of CPM are not yet well established, but they are believed to be a consequence of blood–brain barrier disruption secondary to osmotic stress, leading to demyelination and oligodendrocyte apoptosis.4
Ashrafian and Davey5 have suggested that aetiology is multifactorial and that the syndrome is more likely to present in patients with conditions predisposing to deficiencies in the supply of energy to neurons and glial cells. They also observed that patients with slow correction of hyponatraemia may develop CPM if electrolytic alterations occur during a state of energy deprivation, such as chronic alcoholism or liver disease. These authors report that alcoholic and cirrhotic patients, as well as patients with hypoglycaemia due to other causes, may lack a plentiful supply of glucose or glycogen to glial cells, which is necessary to maintain the activity of the Na+/K+-ATPase pump, the mechanism responsible for electrolyte transport in the brain.5 Subclinical thiamine deficiency may also exacerbate this situation because it decreases glucose uptake in the brain and diminishes the available sources of energy for glial cells. Neurons may release glutamate and other excitatory molecules in response to osmotic stress, increasing intracellular calcium levels. The latter stimulus would lead to cell apoptosis, which these authors suggest may be the final mechanism of CPM.5
Clinical manifestations are very heterogeneous, ranging from asymptomatic to severe neurological disorders such as dysphagia, dysarthria, quadriparesis, and coma; neuropsychiatric disorders including anxiety, emotional lability, apathy, mutism, agitation, and disinhibition; and such movement disorders as parkinsonism and dystonia.3,6 The most frequent initial manifestation is encephalopathy.2 Pontine/extrapontine lesions may occur independently or simultaneously, with extrapontine lesions (either in isolation or in combination with pontine lesions) being less frequent.6 Full recovery has even been reported in patients with severe neurological symptoms.2,3
The gold standard for diagnosis is brain MRI, which is reported to detect even asymptomatic cases, improving outcomes.2 It should be noted that radiological findings may not be observed in the first week after symptom onset, and the MRI scan should be repeated when there is a high level of suspicion.6
Early treatment with dexamethasone after rapid correction of hyponatraemia has been tested in animals, with excellent clinical results and improved prognosis, due to the capacity of dexamethasone to regulate and prevent damage to the blood–brain barrier and to decrease microglial cytokine release.7 However, no controlled trials have been performed in humans to date.
Our case is of special relevance due to several key observations: progression was subacute, isolated hyperglycaemia was the main trigger factor, and clinical symptoms fully resolved after achieving good metabolic control in a patient with cirrhosis as trigger factor.2 Most of the reported cases of CPM occur in the context of hyperglycaemia with concomitant ketoacidosis, abnormal sodium levels, or after treatment for hyperosmolar hyperglycaemic state; less frequently,8–10 onset of CPM is secondary to isolated hyperglycaemia.8,11,12 Therefore, our case would support the hypothesis that fluctuations in osmolarity and even hyperglycaemia itself may act as a trigger factor in the aetiopathogenesis of osmotic demyelination syndrome.
Please cite this article as: Corps Fernández D, Terrero Carpio R, Escolar Escamilla E, Pinel González A. Mielinólisis central pontina de curso subagudo secundario a hiperglucemias. Neurología. 2020. https://doi.org/10.1016/j.nrl.2017.09.009
