




Hereditary spastic paraparesis is a genetically and phenotypically heterogeneous group of neurodegenerative diseases characterised by spasticity and progressive weakness of the lower limbs. Inheritance may be autosomal dominant, autosomal recessive, or X-linked. To date, 72 different types have been described, and clearly defined causal mutations have not been identified for all of them.
We present the case of a 7-year-old boy with intellectual disability, autosomal dominant 9 associated with a heterozygous pathogenic variant of the kinesin family member 1A (KIF1A) gene (missense mutation, Chr2:241724479 C>T, exon 7, p.Arg216His).
The patient was first attended at the age of 3 due to a degree of psychomotor retardation: head control at 2.5 months, sitting unaided at 10 months, first two-syllable words at 13-16 months, and walking at 21 months. The patient was already attending school, but displayed slower learning than his peers, with poorer speech (although comprehension was preserved). His clinical history included birth by Caesarean section at 38 weeks of gestation, with an Apgar score of 10/10. Pregnancy and the neonatal period were uneventful (normal results in the endocrine and metabolic tests and normal growth). The parents were non-consanguineous.
Physical examination revealed hyperreflexia in the lower limbs (especially in the right side of the body), with bilateral extensor plantar reflexes and spastic gait. We observed no alterations in motor strength (minimally increased muscle tone in the right lower limb) or sensitivity; the patient did not present dysmetria.
He was also diagnosed with neurogenic bladder. The ophthalmology and otorhinolaryngology departments also assessed the patient, finding no alterations besides astigmatism in both eyes.
Given the suspicion of spastic paraparesis possibly of genetic cause or secondary to cerebral palsy, we requested a brain and spinal cord MRI study, testing for fragile X syndrome, and karyotyping; all yielded normal results. Levels of pristanic acid, phytanic acid, and very-long-chain fatty acids were also normal. Laboratory analysis showed creatine kinase level of 189U/L, vitamin B12 level of 603pg/mL, vitamin D level of 19ng/mL, vitamin E level of 12.2μg/mL, and no deamidated gliadin or transaminase antibodies.
We also requested somatosensory, auditory, and visual evoked potentials, which revealed normal results (only a slight bilateral increase in latency of P100, probably associated with refractive errors), as well as an electroneurography study, which revealed axonal sensory polyneuropathy of moderate intensity. A follow-up brain MRI scan showed discreet cerebellar atrophy (vermis and hemispheres) (Fig. 1), with enlarged cisternal spaces, which was not detected in an MRI performed 3 years before.

Considering these results, we requested an exome sequencing study to identify genomic variants of 122 genes associated with spastic paraplegia and ataxia; a heterozygous pathogenic variant of the KIF1A gene was detected. A genetic study of the parents revealed that they were not carriers of the mutation.
Gait has improved, as have fine motor skills, although the patient shows impaired attention and writing difficulties. The patient has occasionally reported discomfort in the fingers when performing physical exercise. He acquired sphincter control at the age of 6 years and continued to be followed up by the nephrology department.
The patient is currently attending school, and although he needs significant support he is able to read and write. We should underscore that academic performance improved significantly after prescription of methylphenidate.
The AmpliSeq™ Exome panel (Life Technologies) was used for library preparation. This technique captures > 97% of consensus coding sequences (> 19000 genes, > 198000 exons, > 85% of alterations responsible for genetic disease) and adjacent splice sites (5bp). The panel is approximately 33Mb in size and comprises a total of 293903 amplicons. Library sequencing was performed using the Ion Proton™ next-generation sequencing system (Life Technologies). The sequences obtained were aligned against the reference genome (build 37 of the Hg19 genome) using the TMAP and Ion-Alignment packages of the Torrent Suite software. After alignment and filtering according to specific quality criteria, the sequences were analysed with the Variant Caller tool to identify nucleotide variations with respect to the reference genome. Variant annotation was performed using the latest available version of Ion Reporter™ (Life Technologies), which, in the case of trio analysis, enables the annotation of proband variants by genetic category. Our analysis aimed to identify variants located in exons and splice sites that caused protein-level modifications (missense or nonsense mutations and nucleotide insertions, deletions, or indels) and were detected in over 40% of reads. We evaluated the list of variants identified against database information on previously described polymorphisms (http://www.ncbi.nlm.nih.gov/SNP/, http://www.1000genomes.org, and http://evs.gs.washington.edu/EVS) to identify benign variants common in the general population that are not associated with diseases (variants present in at least 1% of the population).
We also estimated the functional effect of the genomic variations classified as pathogenic using the 7 prediction systems (SIFT, PROVEAN, PolyPhen2, MutationTaster, MutationAssessor, LRT, and FATHMM) included in the ALAMUT (http://www.interactivebiosoftware.com) and ANNOVAR (http://www.openbioinformatics.org/annovar/) analysis packages. Finally, we assessed the association between the mutations identified and OMIM syndromes.
The c.647G>A variant of KIF1A was identified by massive sequencing in a study of 122 genes associated with spastic paraparesis and ataxia. The mean number of reads was 90. Mean read depth of the amplicons included in the study was below 10× in 4.4% of the sequenced amplicons.
The KIF1A variant was confirmed by direct Sanger sequencing. The variant was detected in heterozygosis.
The c.647G>A variant of KIF1A (p.Arg216His) is a missense mutation affecting the protein motor domain, and has been registered as a pathogenic variant in the HGMD database (accession number: CM157166). It results in the c.647G>A nucleotide change (NM_001244008.1) in exon 7 of the KIF1A gene, including 49 exons and located on cytoband 2q37.3. It is therefore an exonic variant. This change provokes the substitution of arginine with histidine at position 216 of the protein (p.Arg216His), causing a missense mutation.
This variant is registered on the dbSNP database, with identification number rs672601368, with no associated allele frequency. The bioinformatic analysis systems used for predicting the effects of the mutation suggested that the variant was deleterious in 6 out of 7 cases (PROVEAN, SIFT, PolyPhen2, MutationTaster, MutationAssessor, and FATHMM).4–9
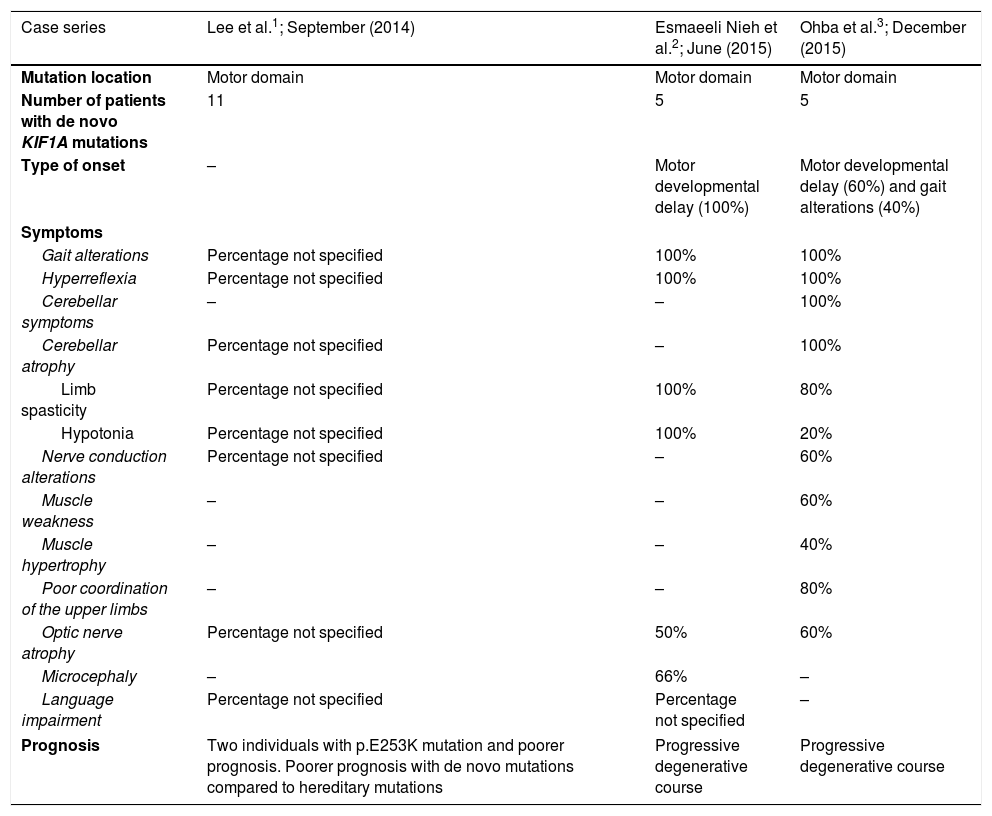
The variant has previously been described in a 16-year-old male patient with peripheral neuropathy, spastic paraparesis, and motor and language developmental delay, among other clinical characteristics. The patient presented 2 de novo variants: the KIF1A c.647G>A variant and a NID1 variant. In this patient, clinical symptoms are thought to be mainly explained by the KIF1A mutation. The presence of de novo missense mutations in the motor domain of the KIF1A gene has previously been associated with clinical symptoms characterised by intellectual disability, spastic paraparesis, axonal neuropathy, and cerebellar atrophy, generally leading to progressive impairment with poor progression (Table 1).10–13
Summary of clinical and genetic characteristics of patients with de novo KIF1A mutations reported in the literature.
| Case series | Lee et al.1; September (2014) | Esmaeeli Nieh et al.2; June (2015) | Ohba et al.3; December (2015) |
|---|---|---|---|
| Mutation location | Motor domain | Motor domain | Motor domain |
| Number of patients with de novo KIF1A mutations | 11 | 5 | 5 |
| Type of onset | – | Motor developmental delay (100%) | Motor developmental delay (60%) and gait alterations (40%) |
| Symptoms | |||
| Gait alterations | Percentage not specified | 100% | 100% |
| Hyperreflexia | Percentage not specified | 100% | 100% |
| Cerebellar symptoms | – | – | 100% |
| Cerebellar atrophy | Percentage not specified | – | 100% |
| Limb spasticity | Percentage not specified | 100% | 80% |
| Hypotonia | Percentage not specified | 100% | 20% |
| Nerve conduction alterations | Percentage not specified | – | 60% |
| Muscle weakness | – | – | 60% |
| Muscle hypertrophy | – | – | 40% |
| Poor coordination of the upper limbs | – | – | 80% |
| Optic nerve atrophy | Percentage not specified | 50% | 60% |
| Microcephaly | – | 66% | – |
| Language impairment | Percentage not specified | Percentage not specified | – |
| Prognosis | Two individuals with p.E253K mutation and poorer prognosis. Poorer prognosis with de novo mutations compared to hereditary mutations | Progressive degenerative course | Progressive degenerative course |
However, our patient displays a degree of clinical improvement in some physical and cognitive areas.
The early use of exome sequencing in diagnosis may save time and resources.
Please cite this article as: Urtiaga Valle S, Fournier Gil B, Ramiro León MS, Martínez Menéndez B. Utilidad del exoma en el estudio de la paraparesia espástica y la atrofia cerebelosa: mutación de novo en el gen KIF1A, una nueva esperanza pronóstica. Neurología. 2020;35:535–538.
