



Genomic studies have identified numerous genetic variants associated with susceptibility to multiple sclerosis (MS); however, each one explains only a small percentage of the risk of developing the disease. These variants are located in genes involved in specific pathways, which supports the hypothesis that the risk of developing MS may be linked to alterations in these pathways, rather than in specific genes. We analyzed the role of the TNFRSF1A gene, which encodes one of the TNF-α receptors involved in a signaling pathway previously linked to autoimmune disease.
MethodsWe included 138 individuals from 23 families including at least 2 members with MS, and analyzed the presence of exonic variants of TNFRSF1A through whole-exome sequencing. We also conducted a functional study to analyze the pathogenic mechanism of variant rs4149584 (-g.6442643C > G, NM_001065.4:c.362 G > A, R92Q) by plasmid transfection into human oligodendroglioma (HOG) cells, which behave like oligodendrocyte lineage cells; protein labeling was used to locate the protein within cells. We also analyzed the ability of transfected HOG cells to proliferate and differentiate into oligodendrocytes.
ResultsVariant rs4149584 was found in 2 patients with MS (3.85%), one patient with another autoimmune disease (7.6%), and in 5 unaffected individuals (7.46%). The 2 patients with MS and variant rs4149584 were homozygous carriers and belonged to the same family, whereas the remaining individuals presented the variant in heterozygosis. The study of HOG cells transfected with the mutation showed that the protein does not reach the cell membrane, but rather accumulates in the cytoplasm, particularly in the endoplasmic reticulum and near the nucleus; this suggests that, in the cells presenting the mutation, TNFRSF1 does not act as a transmembrane protein, which may alter its signaling pathway. The study of cell proliferation and differentiation found that transfected cells continue to be able to differentiate into oligodendrocytes and are probably still capable of producing myelin, although they present a lower rate of proliferation than wild-type cells.
ConclusionsVariant rs4149584 is associated with risk of developing MS. We analyzed its functional role in oligodendrocyte lineage cells and found an association with MS in homozygous carriers. However, the associated molecular alterations do not influence the differentiation into oligodendrocytes; we were therefore unable to confirm whether this variant alone is pathogenic in MS, at least in heterozygosis.
En estudios genómicos se han identificado numerosas variantes genéticas asociadas con la susceptibilidad a la esclerosis múltiple (EM); sin embargo, cada una de ellas explica solo un pequeño porcentaje del riesgo de desarrollar la enfermedad. Estas variantes se localizan en genes implicados en vías específicas, lo que apoya la hipótesis de que el riesgo de desarrollar EM puede estar relacionado con alteraciones en estas vías, más que en genes específicos. Analizamos el papel del gen TNFRSF1A, que codifica para uno de los receptores de TNF-α implicados en una vía de señalización que ha sido previamente relacionada con enfermedades autoinmunes.
MétodosIncluimos a 138 individos de 23 familias que incluían al menos 2 miembros con EM y analizamos la presencia de variantes exónicas de TNFRSF1A mediante la secuenciación del exoma completo. También realizamos un estudio funcional para analizar el mecanismo patogénico de la variante rs4149584 (-g.6442643C > G, NM_001065.4:c.362 G > A,R92Q) por transfección plasmídica en células de oligodendroglioma humano (HOG), las cuales se comportan como células de linaje oligodendrocitario; se utilizó marcaje de proteínas para localizar la proteína dentro de las células. También analizamos la capacidad de las células HOG transfectadas para proliferar y diferenciarse en oligodendrocitos.
ResultadosLa variante rs4149584 se encontró en 2 pacientes con EM (3,85 %), un paciente con otra enfermedad autoinmune (7,6 %) y en 5 individuos no afectados (7,46 %). Los 2 pacientes con EM y variante rs4149584 eran portadores homocigotos y pertenecían a la misma familia, mientras que los individuos restantes presentaron la variante en heterocigosis. El estudio de células HOG transfectadas con la mutación mostró que la proteína no llega a la membrana celular, sino que se acumula en el citoplasma, particularmente en el retículo endoplásmico y cerca del núcleo; esto sugiere que, en las células que presentan la mutación, TNFRSF1 no actúa como una proteína transmembrana, lo que puede alterar su vía de señalización. El estudio de proliferación y diferenciación celular encontró que las células transfectadas siguen siendo capaces de diferenciarse en oligodendrocitos y, probablemente, todavía sean capaces de producir mielina, sin embargo, presentan una menor tasa de proliferación que las células silvestres.
ConclusionesLa variante rs4149584 está asociada con el riesgo de desarrollar EM. Analizamos su papel funcional en células de linaje oligodendrocitario y encontramos una asociación con la EM en portadores homocigóticos. Sin embargo, las alteraciones moleculares asociadas no influyen en la diferenciación a oligodendrocitos; por lo tanto, no hemos podido confirmar si esta variante por sí sola es patógena en la EM, al menos en la heterocigosis.
Different experimental studies have suggested that the TNFRSF1A receptor may play a pathogenic role in multiple sclerosis (MS).1,2 Other studies have described an association between MS and 2 variants of the TNFRSF1A gene, rs1800693 and rs4149584 (p.Arg121Glu (R121Q, also known as R92Q)3; other variants of the gene have also been found in patients with other autoimmune diseases (AID).4 Several studies have focused on the association between MS and rs18006935,6; this variant is known to lack exon 6,7 resulting in a truncated isoform (Δ6-TNFR1) that lacks the extracellular carboxy-terminal portion and therefore presents a functional deficit. Less information is available on the other variant, rs4149584, a polymorphism located in exon 4 that causes an arginine-to-glutamine substitution.8 This R92Q substitution is also the most frequent genetic defect in patients with TNF receptor–associated periodic syndrome (TRAPS), an autoinflammatory disorder characterized by recurrent episodes of fever, myalgia, arthralgia, skin eruptions, abdominal pain, and conjunctivitis; however, this is a low-penetrance mutation associated with reduced up-regulation of inflammatory genes.9
TRAPS is sometimes associated with CNS involvement10,11; furthermore, cases have been reported of adults with MS and TNFRSF1A mutations who present symptoms compatible with TRAPS.12,13 However, R92Q is a low-frequency variant in sporadic cases; therefore, insufficient evidence is available to establish a clear association with the disease, as it has been observed both in patients with MS and in unaffected individuals.14,15 The functional mechanism by which this variant increases the risk of MS is unknown, although it has been suggested that R92Q modifies the contact area between TNF-α and its receptor, increasing electrostatic interactions and interaction with the ligand, which may modify the receptor’s signaling pathway.16
An association has been described between variant rs4149584 and variants of the MEFV gene, associated with familial Mediterranean fever (FMF), a condition with similar clinical features to TRAPS (headache, fever, myalgia, chest or abdominal pain, arthralgia, fatigue, and skin eruption). The protein encoded by this gene is involved in the secretion of the pro-inflammatory cytokine IL-1β, which has been linked to the development of MS. Cases have been reported of FMF associated with CNS involvement17,18 and of patients with both MS and FMF.19–21 It has been suggested that FMF may worsen pre-existing MS,19 and the literature describes a possible association between FMF and MS.21 It has also been hypothesized that mutations in the MEFV gene may be associated with MS22–24 or may influence the secondary effects of interferon beta,25 disease severity,26 and the development of childhood-onset MS.27
Most studies showing an association between variant rs4149584 and MS included heterozygous carriers; however, it is unclear whether homozygosity for the mutation may have a greater impact on the development of MS. This is particularly interesting given that the mutation has been found in homozygosity in 2 patients with TRAPS from 2 unrelated families.28 We describe a family in which homozygosity for variant rs4149584 is associated with MS; the study was conducted in a cohort of patients with familial MS and their relatives, and analyzed the presence of variants in the TNFRSF1A gene. We also studied the functional consequences of polymorphism rs4149584 in an in vitro study of oligodendrocyte lineage cells with a view to analyzing its potential pathogenicity.
MethodsWe included 138 individuals from 23 families with at least 2 patients diagnosed with MS according to the McDonald criteria.29 The study protocol included a specifically-designed questionnaire to assess patients’ personal and family history of neurological, systemic, and autoimmune diseases. Patients also completed a questionnaire aimed at ruling out a potential association with FMF or TRAPS. History of AID was defined according to a modified version of the list of diseases included in the American Autoimmune Related Diseases Association, published elsewhere.30 We also recorded demographic and clinical variables, sex, age at onset, disease progression time, and clinical form (relapsing-remitting, primary-progressive, or secondary-progressive). We gathered information on family members by interviewing them directly; families were classified according to the types of family (type A or type B) defined in a previous study. The free software Genial Pedigree Draw (http://app.pedigreedraw.com) was used to generate pedigrees for each family included.
Whole-exome sequencingBlood samples were gathered from all 138 participants, who had signed informed consent forms. DNA was extracted using the MagNA Pure System (Roche Molecular Systems, Inc.); DNA concentration and purity were evaluated with Qubit™ 2.0 and NanoDrop (Thermo Fisher Scientific, Inc.), respectively. The library was prepared with the Ion AmpliSeq™ Exome Kit (Thermo Fisher Scientific, Inc.), covering > 97% of consensus coding sequences (> 19 000 genes, > 198 000 exons, > 85% alterations responsible for genetic diseases) and adjacent splice sites (5 bp). The panel level is approximately 58 Mb in size and distributed over 293 903 amplicons. To achieve optimal coverage uniformity, DNA libraries were quantified by qPCR and subsequently prepared and enriched with the Ion Chef™ System. The Ion Proton System for next-generation sequencing (Thermo Fisher Scientific, Inc.) achieved coverage of over 90% amplicons with at least 20 reads and a mean coverage depth of > 100 reads in all families. We used the Torrent Mapping Alignment Program to align the sequences obtained to the reference genome (Genome Reference Consortium human genome 19, build 37). Sequences were subsequently filtered according to specific and recommended quality criteria and analyzed using the Variant Caller tool to identify nucleotide variations as compared to the reference genome. All identified variants were annotated using the latest available version of the Ion Reporter™ software (Thermo Fisher Scientific, Inc.). We aimed to identify single-nucleotide polymorphisms (SNP) and indels located within exons and splice sites of the genes analyzed and causing protein alterations, detected in over 40% of reads.
Observing quality control and filtering, we analyzed coding regions and splice sites of TNFRSF1A and MEFV, and created a list of previously described variants (https://www.ncbi.nlm.nih.gov/snp/,http://www.1000genomes.org, https://gnomad.broadinstitute.org/, http://evs.gs.washington.edu/EVS), including the name, alternative names, and genomic location of the genes. We excluded variants located in introns, intergenic regions, and untranslated regions; the remaining variants were classified as either synonymous or non-synonymous. For all variants, we associated population frequency (minor allele frequency [MAF] data from GnomAD database (http://gnomad.broadinstitute.org/). In order to determine the potential biological functions of the variants selected, we estimated the functional effect of the genomic variations classified as pathogenic using 7 prediction algorithms (SIFT, PROVEAN, PolyPhen2, MutationTaster, MutationAssessor, LRT, and FATHMM) included in the ALAMUT (http://www.interactive-biosoftware.com) and ANNOVAR (http://www.openbioinformatics.org/annovar/) analysis packages. We used the Combined Annotation Dependent Depletion tool (CADD, version 1.3) to predict damage to protein function (https://cadd.gs.washington.edu/).31,32 According to the CADD scoring criteria, functional variants score ≥ 10, deleterious variants score ≥ 20, and disease causing variants score ≥30. Finally, we reviewed candidate genes mentioned in the literature available on PubMed and included in the Online Mendelian Inheritance in Man catalogue. Variants were described using their dbSNP ID (rs number). The results of the descriptive analysis of variants are expressed as absolute frequencies (%), means (standard deviation), or medians (interquartile range). We used the Kolmogorov-Smirnov normality test to analyze quantitative data, the chi-square test to compare qualitative variables from independent samples, and the Mann-Whitney U test for quantitative variables. Intergroup differences were analyzed with the Kruskal-Wallis H and Dunn post-hoc tests. The More Powerful Quasi-Likelihood Score Test33,34 was used to test associations between patients and controls; this test is used with samples including related individuals. Allele frequencies were analyzed to test for deviations from Hardy-Weinberg equilibrium. The Bonferroni correction was used for multiple comparisons. We analyzed the variants detected in all groups (individuals with MS, individuals with other AID, and unaffected individuals), considering family type. We also calculated odds ratios (OR) with 95% confidence intervals (CI). A pedigree analysis was performed to evaluate the role of the variants identified in each pedigree, using the criteria established by Sadovnick et al.35: variants present in unaffected family members and not observed in at least 2 family members with MS were considered not to segregate with disease. To this end, both patients with MS and patients with MS plus other AIDs were regarded as cases. Bioinformatic filtering detected synonymous exonic variants, which were not specifically analyzed due to low pathogenic potential; these variants are included in Supplementary Table 2. We also did not analyze intronic variants due to specific capture design limitations. We also detected potential non-synonymous exonic variants that were not listed in the library used, and were therefore considered of uncertain significance. The variants detected were included in a free-access database (European Genome-phenome Archive; https://ega-archive.org/datasets/EGAD00001005952). The presence of variant rs4149584 was confirmed by Sanger sequencing.
Cell culturesWe used a cell line derived from human oligodendroglioma (HOG) (SCC163, Merck). HOG cells were cultured in Dulbecco’s Modified Eagle Medium 1× (DMEM; 1885-023, Gibco) supplemented with 10% fetal bovine serum (FBS; 10500064, Gibco) and antibiotic-antimycotic solution 100× (15240062, Gibco), and were incubated at 37 °C in a humidified atmosphere with 5% CO2. We passaged cells twice per week using TrypLE Express Enzyme (1×) (12604013, Gibco) to detach them from the surface of the flask. HOG cells constitute a useful model for the study of signal transduction pathways of oligodendrocyte metabolism and the role of these cells in neurodegenerative disease.
Plasmid constructionTo enable expression of the mutant protein into cells, we designed expression vectors containing either the wild-type TNFRSF1A gene or the gene with the rs4149584 polymorphism. The pEGFP commercial vector was used as a template, and the P2A and c-myc labels were added. The resulting constructions were called pcDNA-TNFRSF1A_WT_p2AGFP (plasmid containing the wild-type gene) and pcDNA-TNFRSF1A_R92Q_p2AGFP (plasmid containing the mutant gene). Plasmids were synthesized, amplified, and validated by BioAssays, S.L. (Madrid, Spain).
Cell transfectionHOG cells were placed in a 24-well plate with coverslips (∅ 12 mm, #1) coated with poly-l-lysine (P4707 Sigma-Aldrich); 24 hours later, they were transfected with the previously described plasmids using the TransIT®-2020 transfection reagent (MIR 5400, Mirus), according to the manufacturer’s instructions. For a 24-well plate, cells in each well were cotransfected with 1 µg of the plasmid containing either the wild-type gene or the mutant gene, and 2 µL TransIT®-2020 transfection reagent plus 10 µL serum-free medium was added. Cells were harvested 48 hours after transfection and analyzed by immunofluorescence with a confocal microscope.
ImmunocytochemistryWe performed an immunocytochemical analysis of cells expressing GFP (wild-type and mutant), which was part of the plasmid construction, to evaluate the colocalization and location of the transfected protein within cells. Culture cells were fixed in 4% paraformaldehyde (PFA) with 30% sucrose solution for 30 minutes at 37 °C. Cultures were preincubated for one hour in blocking solution (10% goat serum, 0.1% Triton X-100, BSA) and subsequently incubated overnight at 4 °C with the appropriate primary antibody (rabbit polyclonal anti–GRP78 BiP antibody [1:500; Abcam] or wheat germ agglutinin [W21404-WGA Alexa Fluor® 633 conjugate, Invitrogen]), before mounting on glass slides with FluorSave (Calbiochem). GRP78 BiP promotes the assembly of multimeric protein complexes in the endoplasmic reticulum and is involved in the correct folding of proteins and the degradation of misfolded proteins via its interaction with DNAJC10. Wheat germ agglutinin selectively binds to N-acetylglucosaminyl and N-acetylneuraminic acid residues, used with the classic cell membrane dye. Analyses were performed with an Olympus AF-1000 confocal microscope at 40× or 63× magnification; images were acquired in individual (sequential) channels to avoid interferences between fluorescence emission/excitation spectra. Three-dimensional reconstructions were performed for colocalization analysis (Pearson correlation coefficient). For statistical analysis, up to 4 coverslips from 2 independent experiments were counted for each condition, using an Olympus confocal microscope at 40× or 63× magnification. More than 150 transfected cells were counted per coverslip. We conducted a statistical analysis to determine the percentage of each phenotype present in the cells transfected with each plasmid.
Study of proliferation and differentiation of cells with the mutant plasmidBefore transfection, HOG cells were incubated in DMEM culture medium (DMEM 1×, glutamine 2 mM, 10% FBS, and 1% penicillin-streptomycin antibiotic solution) in the dark, at 37 °C with 5% CO2 and 95% relative humidity; these cells were identified by the pcDNA3.2-P2A-GFP plasmid as empty, wild-type, or mutant. The vector was constructed according to BioAssays’ specifications. A total of 80 000 cells/well were incubated for 24 hours using a P6 chamber slide and the culture medium previously described. After 24 hours, the culture medium was changed for serum-free medium, and the cells were transfected with serum-free DMEM 1x, TransIT®-2020 transfection reagent, and the plasmid (the latter 2 at a 4:1 proportion) and incubated at 37 °C and 5% CO2 for 48 hours. The serum-free medium was subsequently replaced with oligodendrocyte precursor cell differentiation medium (OPCDM; #1631, ScienCell Research Laboratories), supplemented with oligodendrocyte precursor cell differentiation supplement (OPCDS; #1672, ScienCell Research Laboratories), as recommended by the manufacturer.
Cells were then fixed with 2.5% PFA; to determine the degree of maturation of transfected cells (empty, wild-type, or mutant plasmid), immunocytochemical staining was performed with oligodendrocyte markers (early and mature oligodendrocytes; ab7474, Abcam), anti–myelin basic protein antibodies (AB5864, Millipore), myelin PLP antibodies (NBP1-87781, Novus Biologicals), and anti-CNPase antibodies (mature myelinated oligodendrocytes; ab44726, Abcam). After incubation with primary antibodies, the cells were incubated with the appropriate secondary antibodies (Alexa Fluor goat anti-mouse or anti-rabbit secondary antibodies). DRAQ5 was used for cell nucleus staining. Immunocytochemistry was performed with an Olympus AF-1000 confocal microscope. We analyzed at least 250 GFP-labeled cells and compared the expression of the marker corresponding to each stage of oligodendrocyte maturation between the 3 groups. We also calculated the proliferation rate in another sample of HOG cells cultured under similar conditions, to analyze whether the mutation may interfere in oligodendroglial maturation/differentiation; confocal microscopy was used to determine the total number of cells and the percentage of viable cells.
Statistical analysisData from the transfection study were analyzed using one-way analysis of variance followed by a Tukey test for multiple comparisons. All values are presented as means ± standard error (SE); statistical significance was set at P < .05. Data from the study of cell proliferation and differentiation are also presented as means ± SE; statistical analysis included analysis of variance and the post-hoc Tukey test. P-values < .05 were considered statistically significant. Analyses were performed using GraphPad Prism 8.
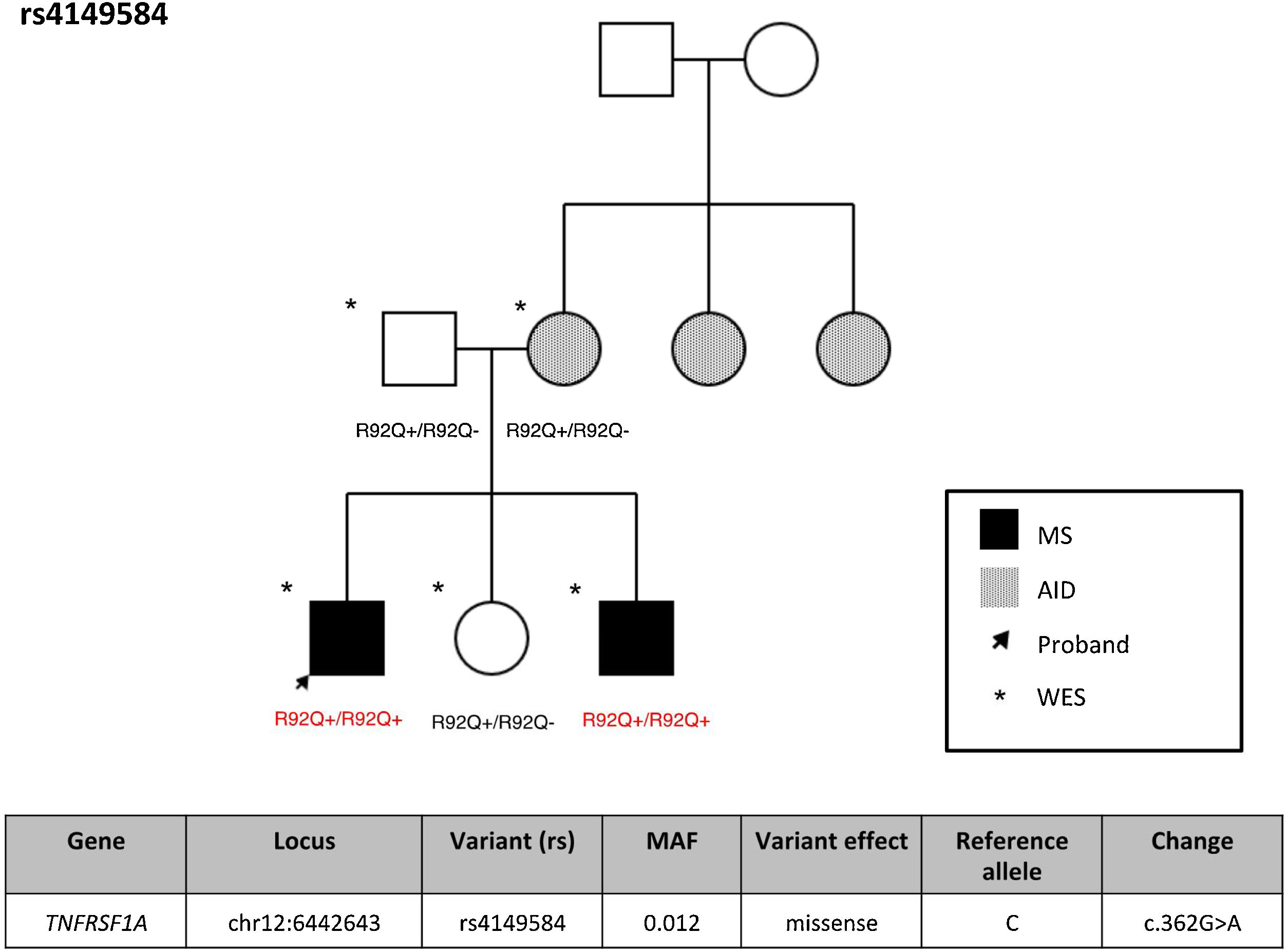
ResultsFrequency of exonic variants of the TNFRSF1A geneSupplementary Table 1 summarizes the demographic characteristics of our cohort of patients with MS. Variant rs4149584 was detected in 2 patients with MS (3.85%), one patient with another AID (7.6%), and 5 unaffected individuals (7.46%). None of the study participants presented TRAPS or FMF. Supplementary Table 2 presents the TNFRS1A variants detected in our cohort, with no significant association with any case. Interestingly, the 2 individuals with MS and variant rs4149584 presented the variant in homozygosis, and belonged to the same family (Fig. 1), whereas the remaining individuals were heterozygous carriers of the variant. In that family, presence of the variant was confirmed by Sanger sequencing (Supplementary Fig. 1).
Changes in transfected cells
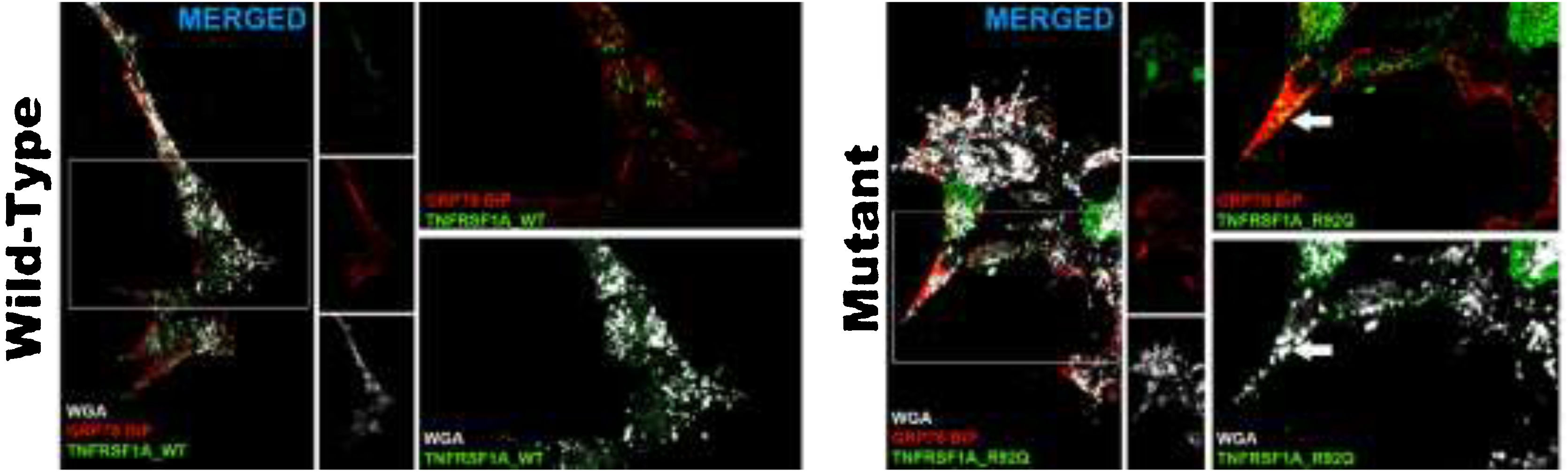
The transfection efficiency of the wild-type plasmid (mean of 78.2 ± 2) was similar to that of the mutant plasmid. The wild-type plasmid presented homogeneous expression, and was expressed in the cell membrane; we observed no autophagic or apoptotic cells or cell detritus expressing GFP. The analysis of cells transfected with rs4149584 revealed marked expression in small intracellular aggregates located in the vicinity of the cell nucleus and occasionally adjacent to the cell membrane (Fig. 2). We used anti–GRP78 BiP antibodies (marker of endoplasmic reticulum) and WGA (marker of cell membranes) to confirm the location of the plasmid transfected into the cell. In cells transfected with the wild-type plasmid, GFP expression colocalized in the cell membrane, and no intracellular aggregates were detected (Fig. 2). In cells transfected with the mutant protein, GFP expression colocalized with the GRP78 BiP marker in intracellular aggregates; we also found that aggregates adjacent to the cell membrane were positive for GRP78 BiP and WGA, displaying colocalization with GFP (Fig. 2). We observed apparently autophagic or karyorrhectic cells, which expressed GFP and the markers of the endoplasmic reticulum and cell membrane.
![Confocal microscopy images of cells transfected with the TNFRSF1A (WT gene) and the TNFRSF1A-R92Q (MT gene). The expression of EGFP coupled to the WT gene and mutant gene in HOG cells is observed, as well as the analysis of the expression of GRP78-BiP (endoplasmic reticulum [ER], protein involved in the correct folding of proteins and degradation of misfolded proteins) and WGA (membrane marking). It is interesting that the expression of TNFRSF1A (WT) was mainly in the cell membrane, whereas TNFRSF1A-R92Q (mutant) expression formed small precipitates in the endoplasmic reticulum (arrows), which are also positive for WGA; these possibly correspond to non-functional/misfolded protein inclusions in the cytoplasm, since GRP78-BiP, also known as HSPA5 (heat shock protein family A, member 5), is considered an essential ER chaperone and a master regulator of ER homeostasis. GRP78 facilitates the folding and assembly of nascent polypeptides, prevents their misfolding and aggregation, targets misfolded proteins for proteasome degradation, and controls signaling for the initiation of the various arms of the unfolded protein response.](https://static.elsevier.es/multimedia/21735808/unassign/S2173580822000876/v1_202208230509/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i85vcbshWVQPfl+0x3NpqwiM8PK/PnqINjlXpgBHub0bcSNvkXab1unOu9IhEgWFGnsBH0Ok3drJE0DQTdcIuO/oayDB857MTnbQOCFiR24s/4DXbU6oYz3BpQo+hdQo8b/JLR1yS5o+LExC7NFKTGYmeV0R4O/+293mwo3kvZ4N9SD2VcOaWRsUaIJF23UGnStfaPlAZgLZ6hrs6MxqsA+KbilH9I/8adx/vL91wWm+aiqYKD+E9IntQ6iV4XZ5h "Confocal microscopy images of cells transfected with the TNFRSF1A (WT gene) and the TNFRSF1A-R92Q (MT gene). The expression of EGFP coupled to the WT gene and mutant gene in HOG cells is observed, as well as the analysis of the expression of GRP78-BiP (endoplasmic reticulum [ER], protein involved in the correct folding of proteins and degradation of misfolded proteins) and WGA (membrane marking). It is interesting that the expression of TNFRSF1A (WT) was mainly in the cell membrane, whereas TNFRSF1A-R92Q (mutant) expression formed small precipitates in the endoplasmic reticulum (arrows), which are also positive for WGA; these possibly correspond to non-functional/misfolded protein inclusions in the cytoplasm, since GRP78-BiP, also known as HSPA5 (heat shock protein family A, member 5), is considered an essential ER chaperone and a master regulator of ER homeostasis. GRP78 facilitates the folding and assembly of nascent polypeptides, prevents their misfolding and aggregation, targets misfolded proteins for proteasome degradation, and controls signaling for the initiation of the various arms of the unfolded protein response.")
Confocal microscopy images of cells transfected with the TNFRSF1A (WT gene) and the TNFRSF1A-R92Q (MT gene). The expression of EGFP coupled to the WT gene and mutant gene in HOG cells is observed, as well as the analysis of the expression of GRP78-BiP (endoplasmic reticulum [ER], protein involved in the correct folding of proteins and degradation of misfolded proteins) and WGA (membrane marking). It is interesting that the expression of TNFRSF1A (WT) was mainly in the cell membrane, whereas TNFRSF1A-R92Q (mutant) expression formed small precipitates in the endoplasmic reticulum (arrows), which are also positive for WGA; these possibly correspond to non-functional/misfolded protein inclusions in the cytoplasm, since GRP78-BiP, also known as HSPA5 (heat shock protein family A, member 5), is considered an essential ER chaperone and a master regulator of ER homeostasis. GRP78 facilitates the folding and assembly of nascent polypeptides, prevents their misfolding and aggregation, targets misfolded proteins for proteasome degradation, and controls signaling for the initiation of the various arms of the unfolded protein response.
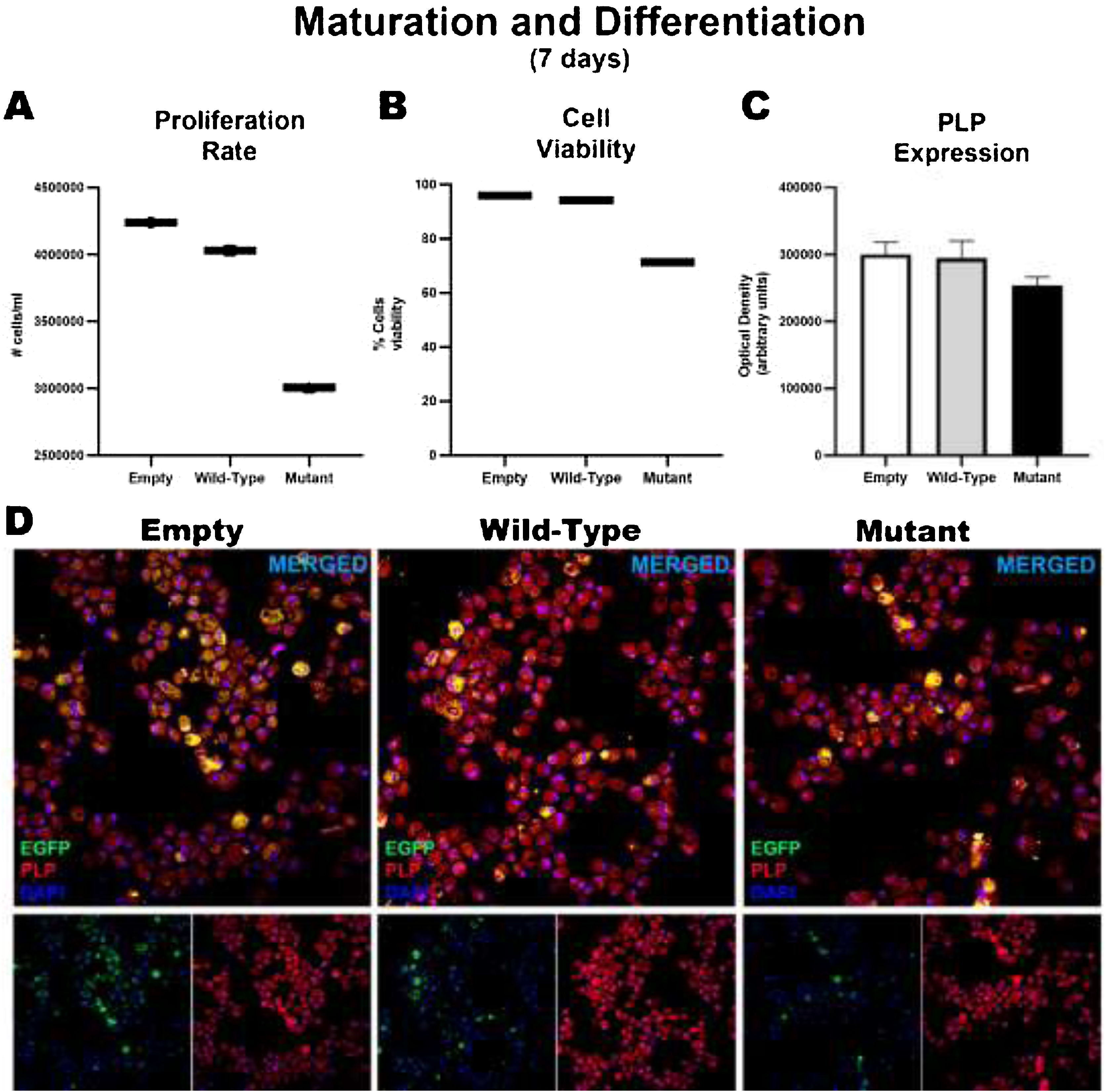
After 7 days of maturation and differentiation, and before transfection, cells showed significant differences in the proliferation rate (empty: 4239369 ± 126469; wild-type: 4030378 ± 123195; mutant: 306110 ± 176833); these differences were correlated with the percentage of viable cells (empty: 96%; wild-type: 94.33%; mutant: 71.4%). However, no significant intergroup differences were observed in the markers used to analyze the degree of maturation and the generation of oligodendrocytes, or in the marker of early oligodendrocytes. Consequently, no differences were observed between wild-type and mutant cells in the expression of MBP and PLP or in the generation of oligodendrocytes, although we did find significant differences in the proliferation rate (Fig. 3).
 and TNFRSF1A-R92Q (MT gene). Graphs A and B show the proliferation rates and cell viability rates of the cells transfected with the empty (EGFP only reporter), WT, and mutant plasmids. Interestingly, there is no difference between cells transfected with WT and empty plasmids, but when comparing those transfected with the mutant gene, a marked difference is observed in the degree of proliferation and cell viability. But when analyzing whether there were alterations in its maturation (expression of myelin PLP) (C and D), no differences were found in its expression at 7 days.")
Maturation and differentiation of HOG cells transfected with TNFRSF1A (WT gene) and TNFRSF1A-R92Q (MT gene). Graphs A and B show the proliferation rates and cell viability rates of the cells transfected with the empty (EGFP only reporter), WT, and mutant plasmids. Interestingly, there is no difference between cells transfected with WT and empty plasmids, but when comparing those transfected with the mutant gene, a marked difference is observed in the degree of proliferation and cell viability. But when analyzing whether there were alterations in its maturation (expression of myelin PLP) (C and D), no differences were found in its expression at 7 days.
It has been suggested that the TNFRSF1A receptor may influence immunity, myelination, and neurodegeneration. TNFRSF1A knockout mice with experimental autoimmune encephalomyelitis (EAE) display fewer symptoms, less marked demyelination, less marked CD3+ cell infiltration, and lower activation of microglia/macrophages. Antibody-mediated inhibition of TNFRSF1A in mice with EAE results in reduced disease severity and less marked demyelination and neuronal loss, with reduced infiltration of T-cells into CNS tissue.36TNFRSF1A silencing in microglial cultures promotes the expression of genes encoding such factors as granulocyte colony-stimulating factor, IL-10, and IFN-γ.37 Lack of TNFRSF1A does not seem to decrease the pool of oligodendrocyte precursor cells (OPC) and oligodendrocytes and, consequently, also seems not to delay remyelination.38 The TNFRSF1A knockout mouse model of retinal ischemia displays a decrease in neuronal cell death.39TNFRSF1A has also been found to have a pro-inflammatory effect and to reduce cell survival in a nucleus basalis lesion model.40
TNFRSF1A gene variantsVariant rs4149584 (rs4149584 (g.6442643C > G, NM_001065.4:c.362 G > A) was only identified in 9 individuals. This variant has a low MAF; its low prevalence in our series was therefore to be expected. However, it should be noted that in one family, the variant always appeared in homozygosis in individuals with MS, whereas the remaining individuals were heterozygous carriers and did not present the disease; we may therefore hypothesize that the variant is pathogenic only when it appears in homozygosis. To our knowledge, this is the first study to report homozygous patients from the same family, suggesting that biallelic presence of variant rs4149584 may increase the risk of MS, indicating a potential association between the variant and the disease. This effect has previously been described in FMF in 2 families, where the disease presented only in homozygous carriers.28 The authors analyzed the effect of homozygosity for the variant, as compared to heterozygosity, on TNF-α–induced cytokine production, and found an association with higher plasma MCP-1 levels and decreased response to TNF-α.28 However, unlike in the patients included in that study, patients homozygous for the gene in our sample did not present increased production of cytokines, particularly TNF-α and IL-6 (data not shown).
Analysis of HOG cells transfected with the mutationWe performed a cell study with a view to understanding the potential action mechanism of variant rs4149584. HOG cells act as OPCs, differentiating into oligodendrocytes with myelinating capacity.41–44 This is the cell line most frequently used in in vitro studies into myelination, since HOG cells share multiple antigenic markers with OPCs, including Olig2, NG2, GalC, CNPase, and growth factor receptors.45 After proliferation and differentiation, HOG cells express PLP, MBP, and MOG, and have the capacity to produce myelin.46,47 As a result, HOG cells have been used in the study of myelination mechanisms48–52 and to analyze the influence of the immune response on OPCs.53–55 However, given that their origin is different from that of OPCs, HOG cells present certain differences, such as lack of GFAP or glutamine expression56 and a tendency to apoptosis under certain circumstances.57 Due to the similarities between HOG cells and OPCs, the former have frequently been used in in vitro studies with different culture media to promote differentiation,58 with a view to analyzing their role in neurological disease,59,60 as well as in studies into the impact of genetic variants on OPCs.61,62 These cells have also been used in multiple sclerosis research63 and in studies with demyelination models.64 OPCs express both TNFRSF1 and TNFRSF265; therefore, HOG cells with both receptors should constitute a suitable model for these analyses. In this study, HOG cells were transfected with plasmid pcDNA-TNFRSF1A_R92Q_p2AGFP in order to analyze the molecular changes caused by the mutation as compared to cells without the mutation. We also used a marker plasmid (pcDNA-TNFRSF1A_p2AGFP) to detect molecular changes. In control cells, the protein colocalizes with the cell membrane, which is consistent with the fact that TNFRSF1 is a transmembrane protein. However, in the cells transfected with the mutation, although the protein does appear in the cell membrane, it is fundamentally found in cytoplasmic aggregates, particularly in the endoplasmic reticulum and in the vicinity of the cell nucleus. We may therefore hypothesize that cells with the mutation do not present TNFRSF1 as a transmembrane protein, which would compromise the TNFRSF1 signaling pathway. This mechanism is different from that generated by mutation rs1800693, which has been associated with a truncated protein (Δ6-TNFR1) that lacks the extracellular carboxy-terminal portion; in the case of rs4149584, the protein does not reach its functional location for ligand binding. It is also different from the mechanism described in a previous functional study, which suggested that the mutation modifies the contact between the transmembrane protein and TNF-α but increases affinity for the ligand. The accepted functional model for the receptor is that ligand binding in the transmembrane domain causes trimerization, which in turn triggers intracellular signaling. Upon dimerization, the rs4149584 mutant protein adopts a different conformational structure from that of the wild-type form66; this may have an impact on the activation of the Nf-κB signaling pathway,67 which has been associated with AID,68 and on the functioning of the intracellular domain of the protein. Nf-κB inhibition has been associated with neurodegeneration.69 Therefore, the functional impact of this mutation may depend on its effect on Nf-κB signaling and how the protein accumulated in the cytoplasm behaves in that pathway.
Mutant HOG cells can produce myelin in vitro but show lower proliferation ratesThe evidence that TNFRSF1 does not act at the transmembrane level in mutant cells is limited by experimental data, given that not only does the TNFRSF1 receptor knockout mouse model not promote demyelination, but these animals also develop less severe EAE.36,38 Therefore, we designed an experiment aimed at demonstrating whether mutant HOG cells behave similarly to OPCs in terms of myelination capacity (like wild-type HOG cells) or whether they lose this capacity, which would support the hypothesis that the mutation is pathogenic and its association with MS. Our results show that HOG cells transfected with the mutation present myelination capacity, since they express myelin proteins and differentiate into oligodendrocytes, which indicates that the mutation in itself is not associated with impaired remyelination; this is consistent with the results of other studies suggesting that lack of the TNFRSF1 receptor does not decrease remyelination capacity.38 However, as may be expected, these cells proliferate at a lower rate. Neural stem cells express TNFR1 and TNFR2,70 and the lack of these receptors results in a lower number of cells available for differentiation.71
LimitationsThe experimental model used, in which the mutant plasmid was inserted but the receptor was not eliminated, prevents extrapolation of our findings to heterozygous cells, since in homozygous cells the lack of TNFRSF1 protein at the transmembrane level must be complete. Presence of the mutation does not change HOG cell functionality. Therefore, our results do not support the hypothesis of the pathogenicity of this mutation and its potential association with MS, although its pathogenicity in homozygosis cannot be ruled out, as in the 2 cases presented here. This would require silencing of the receptor originally present in the cells. TNFRSF1A was present in nearly all cells, whereas TNFRSF1B appeared only in certain populations of T-cells, endothelial cells, microglia, and specific subtypes of neurons, oligodendrocytes, cardiac myocytes, thymocytes, and mesenchymal stem cells. As a consequence, oligodendrocytes express both TNFRSF1B and TNFRSF1A,72 and both receptors bind to TNF-α73; therefore, in this situation, presence of the TNFRSF1B receptor may compensate for the lack of TNFRSF1A in the cell membrane. The lower proliferation rate of the mutant cells is noteworthy. When the pathogenicity of a mutation is analyzed, it is frequently studied in the context of the pathogenic mechanisms of the disease, with less focus on such factors as clinical presentation and severity; it should be noted, however, that a mutation may not be causal but could still influence other aspects of a disease.74
ConclusionsAlthough TNFRSF1A variants are rare, one of the families participating in our study included 2 members with MS who were heterozygous carriers of mutation rs4149584. Despite this, we were unable to confirm the pathogenicity of this variant by transfecting the mutation into oligodendrocyte lineage cells. However, we did observe that this variant is associated with a decrease in protein transport to the plasma membrane. The presence of the TNFRSF1B receptor in cells involved in MS may prevent this influence; this is not the case in patients with other AID that affect cells not expressing this receptor. As an example, the heterozygous carriers included in our study did not display pro-inflammatory cytokine release, unlike previously reported cases of FMF.
Although future research should analyze the role of rs4149584 when it presents alongside other mutations in the same signaling pathway,75 our results raise questions about whether this polymorphism contributes to susceptibility to MS.
DisclosureThe authors have no conflicts of interest to declare.
Ethics statementThe study was approved by the Clinical Research Ethics Committee at Hospital Clínico San Carlos. All participants gave written informed consent. Data were handled in observance of Spanish legislation on data protection (Organic Law 15/1999 of 13 December). Our study complies with the principles of the Declaration of Helsinki (Recommendations guiding physicians in biomedical research involving human subjects, Helsinki 1964, amended October 2013).
Author contributionsLead researchers: JMG, UGP, JAMG; clinical/genetic study design: VP, JAMG, SA, JMG; patient examination: JAMG, PME, VP, LVB, JMG; family studies: VP, LTF, PME; experimental design: JMG, UGP; plasmid generation: RM; experimental data coordination: VP, LTF, MSBM, UGP; whole-exome sequencing: PM, SA; genetic database: VP, JAMG, LTF, JMG, NVG; data filtering and analysis: LH, LTF, JMG, JAMG; genetic data coordination: JMG, JAMG, UGP; cell experiments: UGP, BSC, LMJ, IS; statistical analysis: JAMG, LH, UGP; analysis of results: LTF, JMG, UGP, JAMG; figures and tables: LTF, DOH UGP, JMG; manuscript drafting: JMG, UGP, JAMG; critical review of the manuscript: all authors.
The authors would like to thank the Spanish Society of Neurology’s Research Operations Office, for assisting with the English-language version of the manuscript, and BioRender (https://biorender.com, July 2022), for the images provided. The authors also wish to thank all the individuals who participated in the study.
The following is Supplementary data to this article:
