




Several studies have analysed the presence of P2RX7 variants in patients with MS, reporting diverging results.
MethodsOur study analyses P2RX7 variants detected through whole-exome sequencing (WES).
ResultsWe analysed P2RX7, P2RX4, and CAMKK2 gene variants detected by whole-exome sequencing in all living members (n = 127) of 21 families including at least 2 individuals with multiple sclerosis. P2RX7 gene polymorphisms previously associated with autoimmune disease. Although no differences were observed between individuals with and without multiple sclerosis, we found greater polymorphism of gain-of-function variants of P2RX7 in families with individuals with multiple sclerosis than in the general population. Copresence of gain-of-function and loss-of-function variants was not observed to reduce the risk of presenting the disease. Three families displayed heterozygous gain-of-function SNPs in patients with multiple sclerosis but not in healthy individuals. We were unable to determine the impact of copresence of P2RX4 and CAMKK2 variants with P2RX7 variants, or the potential effect of the different haplotypes described in the gene. No clinical correlations with other autoimmune diseases were observed in our cohort.
ConclusionsOur results support the hypothesis that the disease is polygenic and point to a previously unknown mechanism of genetic predisposition to familial forms of multiple sclerosis. P2RX7 gene activity can be modified, which suggests the possibility of preventive pharmacological treatments for families including patients with familial multiple sclerosis.
Varios estudios han analizado la presencia de variantes de P2RX7 en pacientes con EM, reportando resultados divergentes.
MétodosNuestro estudio analiza las variantes de P2RX7 detectadas mediante secuenciación del exoma completo (WES).
ResultadosAnalizamos las variantes de los genes P2RX7, P2RX4 y CAMKK2 detectadas por secuenciación del exoma completo en todos los miembros vivos (n = 127) de 21 familias, incluidas al menos 2 personas con esclerosis múltiple. Polimorfismos del gen P2RX7 previamente asociados con enfermedades autoinmunes. Aunque no se observaron diferencias entre individuos con y sin esclerosis múltiple, encontramos mayor polimorfismo de variantes de ganancia de función de P2RX7 en familias con individuos con esclerosis múltiple que en la población general. No se observó que la copresencia de variantes de ganancia de función y pérdida de función redujera el riesgo de presentar la enfermedad. Tres familias mostraron SNP de ganancia de función heterocigotos en pacientes con esclerosis múltiple, pero no en individuos sanos. No pudimos determinar el impacto de la copresencia de las variantes P2RX4 y CAMKK2 con las variantes P2RX7, ni el efecto potencial de los diferentes haplotipos descritos en el gen. No se observaron correlaciones clínicas con otras enfermedades autoinmunes en nuestra cohorte.
ConclusionesNuestros resultados apoyan la hipótesis de que la enfermedad es poligénica y apuntan a un mecanismo previamente desconocido de predisposición genética a formas familiares de esclerosis múltiple. La actividad del gen P2RX7 puede modificarse, lo que sugiere la posibilidad de tratamientos farmacológicos preventivos para familias que incluyen pacientes con esclerosis múltiple familiar.
P2X7 is a trimeric receptor that functions as a nonselective cationic channel permeable to Ca2+, Na+, and K+ in the plasma membrane, activated by extracellular ATP; with prolonged ATP activation, however, the receptor may act as a pore, allowing molecules to cross the cell membrane and triggering a signalling pathway that leads to the release of inflammatory mediators. The receptor also participates in cell proliferation and clearance of intracellular pathogens. Several researchers have suggested that P2X7 receptor function is genetically determined,1 with variants increasing or decreasing its activity.2 The P2X7 receptor participates in a number of immune functions, including ATP release in immune response,3 by mediating inflammasome activation4; release of such cytokines as IL-1β, IL-2, and IL-18; T-cell activation and proliferation; and antigen presentation.5,6 Studies with P2X7 receptor–deficient mice7,8 have shown that the protein is involved in a wide range of immune functions, constituting a potential target for the treatment of autoimmune diseases (AID).9,10 The gene coding for the P2X7 receptor (P2RX7, located on 2q24.31 and containing 13 exons) is highly polymorphic, and multiple single-nucleotide polymorphisms (SNP) potentially altering its function have been detected.11,12 Research into the potential therapeutic usefulness of the gene is therefore limited by genetic variability between patients. Being an AID, multiple sclerosis (MS) may benefit from the potential therapeutic usefulness of P2RX7,13 particularly because the P2X7 receptor is overexpressed in regulatory T cells, which are reported to be involved in the pathogenesis of AIDs.14 However, the P2X7 receptor is expressed not only in immune cells but also in central nervous system cells, such as activated microglia,15 astrocytes,16 oligodendrocytes,17–19 and neurons,20 sparking interest in its usefulness in research into neurodegenerative diseases,21 including Alzheimer disease22 and amyotrophic lateral sclerosis.23
Experimental findings support the P2X7 receptor’s role in the pathogenesis of experimental allergic encephalomyelitis, an experimental model of MS: greater receptor expression contributes to myelin damage, whereas P2X7 receptor blockade protects against myelin loss24–26; a partial decrease in activity of the receptor exacerbates symptoms, however.27 Increased P2X7 receptor expression has been observed in spinal cord microglia in patients with MS28; however, expression decreases in monocytes during the acute stage of MS and experimental allergic encephalomyelitis, and is accompanied by alterations in the P2X7 receptor protein and mRNA.29
Genetic research into MS is mainly based on genome-wide association studies (GWAS), which have identified variants of the major histocompatibility complex, located on the short arm of chromosome 6, and multiple low-risk loci in association with the disease. This has led to the conclusion that MS is a polygenic disease.30 Although some of these findings may be affected by methodological biases,31 there is a need for further research into genes involved in signalling pathways in MS, including variants of the P2RX7 gene. Several studies have analysed the presence of P2RX7 variants in patients with MS,32–35 reporting diverging results. Our study analyses P2RX7 variants detected through whole-exome sequencing (WES).
Material and methodsStudy cohortWe studied all living members of 21 families including at least 2 individuals with MS according to the 2010 McDonald criteria,36 with the total sample comprising 127 individuals. Families were classified into 2 groups according to the criteria established in a previous article by our research group37: type A families, with only one generation affected by MS (12 families; 57.1%), and type B families, with patients with MS in at least 2 different generations (9 families; 42.9%). We reviewed the participants’ histories of AID and categorised them using the classification created by the American Autoimmune Related Diseases Association.38 We also recorded demographic variables, personal history, clinical variables, sex, age at disease onset, disease progression time, and clinical form of MS (relapsing-remitting [RRMS], primary progressive [PPMS], or secondary progressive [SPMS]). The Genial Pedigree Draw software (http://app.pedigreedraw.com) was used to draw the pedigrees of all families included.
Whole-exome sequencing and bioinformatics analysisAll participants signed informed consent forms. WES was performed on blood samples from all 127 participants. DNA was extracted using the MagNA Pure System (Roche Molecular Systems, Inc.). The Qubit™ 2.0 fluorometer and NanoDrop™ spectrophotometer (Thermo Fisher Scientific, Inc.) were used to evaluate DNA concentration and purity. The Ion AmpliSeq™ Exome RDY Panel (Thermo Fisher Scientific, Inc.) was used for library preparation. This technique analyses > 97% of consensus coding sequences (>19 000 genes, > 198 000 exons, > 85% alterations responsible for genetic diseases) and adjacent splice sites (5 bp). The panel is approximately 33 Mb in size and comprises a total of 293 903 amplicons. Libraries were quantified by qPCR and subsequently prepared and enriched using the Ion Chef™ System, which achieves high coverage uniformity. For all the families included, we obtained over 90% coverage for a minimum of 20 reads. For library sequencing, we used a mean coverage depth of > 100 reads using the Ion Proton System for next-generation sequencing (Thermo Fisher Scientific, Inc.), which covers > 90% of amplicons with a minimum of 20 reads. The sequences obtained were aligned against the reference genome (Genome Reference Consortium human genome 19, build 37) using the Torrent Mapping Alignment Program. After alignment, sequences were filtered according to specific quality criteria and analysed using the Variant Caller tool to identify variations in nucleotides as compared to the reference genome. Variant annotation was performed using the latest available version of the Ion Reporter™ software (Thermo Fisher Scientific, Inc.). We aimed to identify SNPs and indels located within exons and splice sites of the genes analysed and causing protein alterations (synonymous variants were excluded), detected in over 40% of reads.
Observing quality control and filtering, we analysed coding regions and splice sites of P2RX4, P2RX7, and CAMKK2, and created a list of previously described variants (http://www.ncbi.nih.gov/SNP/, http://www.1000genomes.org, https://gnomad.broadinstitute.org/, http://evs.gs.washington.edu/EVS). We excluded variants located in introns, intergenic regions, and untranslated regions, and classified the remaining variants as either synonymous or non-synonymous. For all variants, we recorded data on population frequency (minor allele frequency [MAF]) (http://gnomad.broadinstitute.org/). In order to understand the possible biological functions of the variants selected, we estimated the functional effect of the genomic variations classified as pathogenic using 7 prediction algorithms (SIFT, PROVEAN, PolyPhen2, MutationTaster, MutationAssessor, LRT, and
FATHMM) included in the ALAMUT (http://www.interactive-biosoftware.com) and ANNOVAR (http://www.openbioinformatics.org/annovar/) analysis packages. We used the Combined Annotation Dependent Depletion tool (CADD, version 1.3) to predict damage to protein function (https://cadd.gs.washington.edu/).39,40 According to the CADD scoring criteria, functional variants score ≥ 10, with deleterious variants scoring ≥ 20 and disease causal variants scoring ≥ 30. Finally, we reviewed candidate genes in publications on PubMed and the Online Mendelian Inheritance in Man catalogue.
Analysis of variantsVariants were described using the rs number; Table 1 shows changes identified in the nucleotide and amino acid sequences. The results of the descriptive analysis of variants are expressed as absolute frequencies (%), means (standard deviation), or medians (interquartile range). Quantitative data were analysed with the Kolmogorov-Smirnov normality test. The chi-square test was used to compare independent samples with qualitative variables, and the Mann-Whitney U test for quantitative variables. Intergroup differences were analysed using the Kruskal-Wallis H test and the Dunn post-hoc test. The More Powerful Quasi-Likelihood Score Test41,42 was used to test associations between patients and controls; this test is used with samples including related individuals. Allele frequencies were analysed to test for Hardy-Weinberg equilibrium. The Bonferroni correction was used for multiple comparisons. We analysed the variants detected in all groups (individuals with MS, individuals with other AIDs, and healthy individuals), considering family type. We calculated odds ratios (OR) with 95% confidence intervals (CI) to guarantee populations were different. Frequencies were compared between data from our sample and from databases providing population frequencies; data from worldwide and European populations were drawn from the gnomAD browser (gnomad.broadinstitute.org) and data from the Spanish population were obtained from Ensembl (http://www.ensembl.org/Homo_sapiens/Info/Index), for the year 2014.
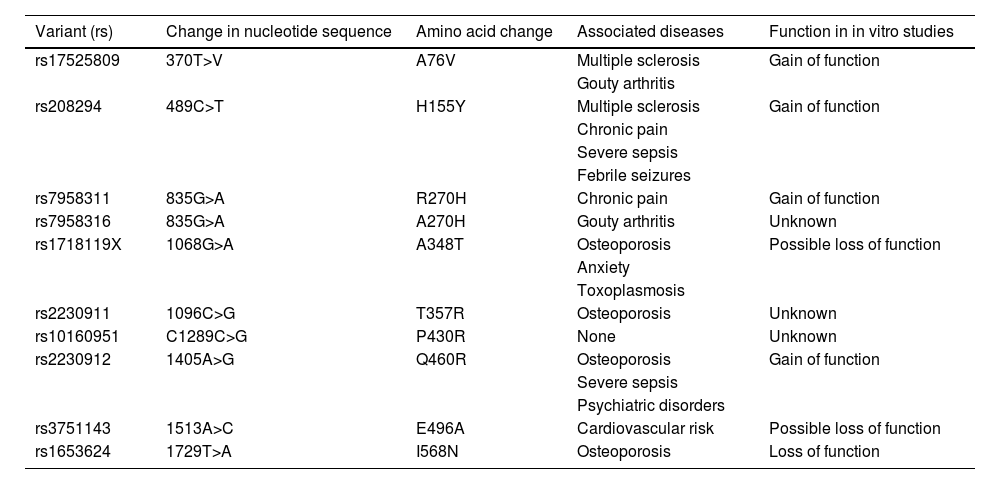
Most frequent P2RX7 variants, classified by rs number. The table provides data on changes in the nucleotide sequence, amino acid changes, associations with diseases or conditions reported in the literature, and gain or loss of function in in vitro studies (adapted from Caseley et al.60).
| Variant (rs) | Change in nucleotide sequence | Amino acid change | Associated diseases | Function in in vitro studies |
|---|---|---|---|---|
| rs17525809 | 370T>V | A76V | Multiple sclerosis | Gain of function |
| Gouty arthritis | ||||
| rs208294 | 489C>T | H155Y | Multiple sclerosis | Gain of function |
| Chronic pain | ||||
| Severe sepsis | ||||
| Febrile seizures | ||||
| rs7958311 | 835G>A | R270H | Chronic pain | Gain of function |
| rs7958316 | 835G>A | A270H | Gouty arthritis | Unknown |
| rs1718119X | 1068G>A | A348T | Osteoporosis | Possible loss of function |
| Anxiety | ||||
| Toxoplasmosis | ||||
| rs2230911 | 1096C>G | T357R | Osteoporosis | Unknown |
| rs10160951 | C1289C>G | P430R | None | Unknown |
| rs2230912 | 1405A>G | Q460R | Osteoporosis | Gain of function |
| Severe sepsis | ||||
| Psychiatric disorders | ||||
| rs3751143 | 1513A>C | E496A | Cardiovascular risk | Possible loss of function |
| rs1653624 | 1729T>A | I568N | Osteoporosis | Loss of function |
A pedigree analysis was performed to evaluate the role of the variants identified in each pedigree, using the criteria established by Sadovnick et al.24: variants present in unaffected family members and not observed in at least 2 family members with MS were considered not to segregate with disease. To this end, both patients with MS and patients with MS and AIDs were regarded as cases. Considering that P2RX7 displays linkage disequilibrium, we aimed to analyse the presence of haplotypes, analysing deviations from Hardy-Weinberg equilibrium and considering the potential associations of alleles described in the literature, both in P2RX743 and in the adjacent loci of P2RX4 and CAMKK2. We manually searched for haplotypes using information published in the literature32,34,44,45 and the Haploview software.
ResultsSynonymous and non-synonymous exonic variantsSupplementary material Table 1 lists the synonymous variants detected, broken down by clinical group (patients with MS, patients with AID, healthy individuals); no significant associations were observed between these variants and MS. The table also compares patients with MS and AIDs against unaffected individuals, and type A against type B families, revealing no significant differences. We also provide a list of non-synonymous variants, which show no significant differences between groups. Additional information on non-synonymous variants is also listed, including presence of homozygous variants, with no relevant findings. The analysis of variant frequency revealed that the degree of polymorphism of P2RX7 for some variants is clearly higher than expected considering MAF. To verify this hypothesis, we compared the frequency of variants in our sample against frequency data from worldwide, European, and Spanish populations: P2RX7 showed markedly greater polymorphism in families with MS than in the general population. Differences were more marked for rs7958311 (0.65; 95% CI, 0.567-0.730 vs 0.24; 95% CI, 0.244-0.247), rs208294 (0.85; 95% CI, 0.778-0.902 vs 0.52; 95% CI, 0.519-0.522), rs2230912 (0.26; 95% CI, 0.191-0.342 vs 0.13; 95% CI, 0.138-0.140), rs1718119 (0.48; 95% CI, 0.402-0.574 vs 0.395, 95% CI, 0.392-0.398), and rs2230911 (0.25; 95% CI, 0.191-0.342 vs 0.139; 95% CI, 0.138-0.140) as compared to worldwide, European, and Spanish populations. Some variants showed differences against the worldwide population but not European or Spanish populations, probably due to sample sizes. Supplementary material Table 1 also shows differences between type A and type B families; the variant rs1718119 is significantly more frequent in type B families (0.698; 95% CI, 0.564-0.804 vs 0.338; 95% CI, 0.240-0.451).
Analysis of exonic variants in pedigreesPedigrees were analysed according to the criteria established by Sadovnick et al.34 The pedigrees of 3 families are particularly interesting (Fig. 1A, B, C). In family 1, rs208294 was only found in patients with MS but not in unaffected family members. This variant (463T>C) was observed in heterozygosis, and has previously been described in association with MS.32 Family 2 included 4 siblings, 2 of whom had MS; the parents did not present the disease. This family showed a variant meeting the Sadovnick criteria, rs7958311, in heterozygosis. Family 3 displayed variants rs7958311 and rs7958316, both of which were observed in heterozygosis in 2 patients with MS and were not observed in the 2 unaffected individuals analysed.
 Family displaying variant rs208294 (463 T>C), which was found in patients with MS but not in unaffected family members; the mutation was present in heterozygosis. B) Pedigree of a family including 4 siblings, 2 of whom have MS, and their parents (the father is deceased), who did not have the disease. This family displays variant rs7958311 (809 G>A) in heterozygosis in the affected individuals only. C) Family displaying variants rs7958311 (809 G>A) and rs7958316 (827 G>A), both of which were present in heterozygosis in 2 patients with MS; the variants were not observed in the 2 unaffected individuals. D) Family showing an association between CAMKK2 variant rs3817190 and P2RX7 variant rs1718119 in 2 patients with MS plus another AID and in one patient with an AID, but not in healthy family members. Arrow: proband; red asterisk: variant; blue arterisk: variant; black: MS; grey: AID.")
Pedigrees of the families meeting the criteria proposed by Sadovnick et al.: variants present in unaffected family members and not observed in at least 2 family members with MS were considered not to segregate with disease. A) Family displaying variant rs208294 (463 T>C), which was found in patients with MS but not in unaffected family members; the mutation was present in heterozygosis. B) Pedigree of a family including 4 siblings, 2 of whom have MS, and their parents (the father is deceased), who did not have the disease. This family displays variant rs7958311 (809 G>A) in heterozygosis in the affected individuals only. C) Family displaying variants rs7958311 (809 G>A) and rs7958316 (827 G>A), both of which were present in heterozygosis in 2 patients with MS; the variants were not observed in the 2 unaffected individuals. D) Family showing an association between CAMKK2 variant rs3817190 and P2RX7 variant rs1718119 in 2 patients with MS plus another AID and in one patient with an AID, but not in healthy family members. Arrow: proband; red asterisk: variant; blue arterisk: variant; black: MS; grey: AID.
Supplementary material table 2 lists the synonymous variants detected in the genes potentially overlapping with P2RX7 (P2RX4 and CAMKK2), broken down by clinical group (patients with MS, patients with AID, healthy individuals); no significant associations were observed between these variants and presence of MS. The table also compares patients with MS or AIDs against unaffected individuals, and type A against type B families, revealing no significant differences. It also provides a list of non- synonymous variants, which show no significant differences, and additional information about non-synonymous variants, including presence of the variants in homozygosis and comparison between type A and type B families, with no remarkable findings. We analysed the presence of haplotypes containing variants of these genes in combination with P2RX7 variants; a P2RX7 variant was associated with a CAMKK2 variant in one family; no associations were observed between P2RX7 and P2RX4 variants. Family 2 shows an association between CAMKK2 variant rs3817190 and P2RX7 variant rs1718119 in 2 patients with MS plus AIDs and in a patient with an AID, but not in healthy family members (Fig. 1D). We also found 2 variants associated with a CAMKK2 mutation linked to MS and meeting the Sadovnick criteria34: rs3817190 and rs28360477.
Clinical characteristics and correlation with P2RX7 variantsThe group of patients with MS included 25 women (54.3%) and 21 men (45.7%); mean age was 46.17 (SD: 18.38) years. The group of patients with AID included 15 women (88.2%) and 2 men (11.8%), with a mean age of 58.24 (11.18) years. The group of unaffected individuals comprised 38 women (59.4%) and 26 men (40.6%), with a mean age of 54.58 (18.99) years. Regarding type of MS, 36 patients (78.3%) had RRMS, 6 (13.0%) had SPMS, and 4 (8.7%) had PPMS, with a mean progression time of 223.26 (SD: 110.45) months. Ten patients with MS (21.7%) also had an AID. Participants were classified by type of family (A or B): 25 patients with MS (54.3%) came from type A families and 21 (45.7%) from type B families, 11 patients with AID (64.7%) came from type A families and 6 (35.3%) from type B families, and 38 unaffected individuals (59.4%) came from type A families and 26 (40.6%) from type B families. Of the patients with MS from type A families, 14 were women (56.0%) and 11 were men (44.0%), with a mean age of 44.80 (7.27) years. Of these patients, 21 (84.0%) had RRMS, 3 (12.0%) had SPMS, and 1 (4.0%) had PPMS. Mean progression time was 219.60 (112.37) months; 4 patients also had an AID. Patients with MS from type B families had a mean age of 47.81 (13.19) years; 11 were women (52.4%) and 10 were men (47.6%). Regarding clinical type of MS, 15 (71.4%) had RRMS, 3 (14.3%) had SPMS, and 3 (14.3%) had PPMS. Mean progression time in this group of patients was 227.62 (110.77) months; 6 patients (28.6%) also had an AID. We also compared the clinical characteristics of MS patients with and without variants rs7958311, rs208294, rs2230912, rs1718119, and/or rs2230911.
We compared the clinical characteristics of MS patients with and without rs1718119, which was found to be significantly more frequent in type B families, to determine whether it was associated with a different clinical profile. The 24 patients with variant rs1718119 had a mean age of 45.12 (SD: 10.31) years; 9 (37.5%) were women and 15
(62.5%) were men. Nineteen (79.2%) had RRMS, 4 (16.7%) had SPMS, and one (4.2%) had PPMS. Mean age at disease onset was 28.42 (9.20) years, mean progression time was 202.87 (SD: 96.22) months, and 6 (25.0%) had another AID in addition to MS. The 22 MS patients without rs1718119 had a mean age of 47.32 (10.58) years; 16 (72.7%) were women and 6 (27.3%) were men. Regarding MS type, 17 patients (77.3%) had RRMS, 2 (9.1%) had SPMS, and 3 (13.6%) had PPMS. Mean age at onset was 26.73 (7.47) years and mean progression time was 245.50 (122.51) months; 4 patients (18.2%) had another AID. No significant differences were observed between groups except for sex: 62% of patients with variant rs1718119 were men, whereas 72.7% of patients without the variant were women. The 41 patients with variant rs208294 had a mean age of 46.49 (10.52) years; 23 (56.1%) were women and 18 (43.9%) were men. Thirty-two (78.0%) had RRMS, 5 (12.2%) had SPMS, and 4 (9.8%) had PPMS. Mean age at disease onset was
27.54 (8.38) years, with a mean progression time of 228.02 (107.93) months; 9 (22.0%) had another AID. The 5 MS patients without rs1718119 had a mean age of 43.60 (9.76) years; 2 (40.0%) were women and 3 (60.0%) were men. Regarding MS type, 4 patients (80.0%) had RRMS and one (20.0%) had SPMS. Mean age at disease onset was 28.20 (9.25) years and mean progression time was 184.20 (136.42) months; one patient (20.0%) had another AID. None of these differences was statistically significant.
To analyse the antagonistic or synergistic effect of different variants presenting in a single patient, we compared the clinical characteristics of MS patients with rs208294 against those with both rs208294 and rs178111, as the latter variant has a loss-of-function effect. We also analysed whether copresence of several variants in a single patient increases the risk of MS. To this end, we compared the frequency of variants rs7958311, rs208294, rs2230912, and rs2230911, analysing whether they present simultaneously in patients with MS, patients with AID, and unaffected individuals. Only a combination of 3 variants showed a stronger association with MS (30.4% vs 18.8% in unaffected individuals), although the difference was not statistically significant.
DiscussionGenetic factors have an impact on susceptibility to MS45–47 and may also influence the course of the disease48; studies analysing these factors should therefore aim to establish clinical correlations.31 SNP linkage analysis, and especially GWAS, has detected a considerable number of loci associated with the disease. However, the alleles detected are neither necessary nor sufficient to cause MS. GWAS do not easily detect rare variants with a potentially significant impact on the disease. While GWAS are based on the hypothesis of common variants associated with a disease and provide information on the risk associated with common genetic variability,49,50 family studies evaluate the effect of a specific variant in individuals with a common genetic background and looking for a Mendelian inheritance model51 and search for genetic factors from a different perspective.50,52 Therefore, information from family studies complements that provided by studies of sporadic cases of MS. One advantage of family studies is the fact that
controls are members of the same family, minimising the impact of such confounders as ethnic and geographical differences. Furthermore, these studies are characterised by greater genetic homogeneity between individuals and allow a better understanding of associated autoimmunity. However, they also have some limitations, including the small size of the samples and the reduced statistical power of analysing the cohort as a whole. Analysing the whole family, as in our study, rather than only trios (an affected individual and 2 first-degree relatives), increases the statistical power of family-based studies.
Description of families including individuals with MS and variants of P2RX7, and association with the diseaseVery few families with cases of MS and variants of P2RX7 have been described. Variant rs200826512 (c.1475G>A; A492L) was first described by Zrzavy et al.35 in a family including 3 individuals with MS; a genetic study performed in 2 of them (a 66-year-old man and his great-nephew) revealed this mutation in heterozygosis. Two unaffected relatives did not have the mutation. The authors support the hypothesis of a very low MAF for this variant. The variant was not observed in any of the 552 patients with MS included in their cohort, which is consistent with the hypothesis of a low allele frequency. We did not detect this variant in any of our families; however, as rs200826512 is in an area that is captured by the WES. Sadovnick et al.34 used the same sequencing method as our own and also failed to detect the variant in their sample. Despite the conclusions drawn by Zrzavy et al., the data do not seem to support an association between rs200826512 and presence of MS, since the rareness of the variant does not necessarily imply that it has a functional impact. Sadovnick et al.34 analysed a cohort of individuals with familial MS. The researchers had previously described a related variant in another study.33 They used the Ion Proton System for 193 patients and 100 controls, identifying 23 non-synonymous variants in P2RX7 and 4 in P2RX4. Nine of the variants described by Sadovnick et al. were also found in our patients: rs17525809, rs208294, rs7958311, rs7958316, rs1718119, rs2230911, rs2230912, rs3751143, and rs16553624. Only 2 of these (rs7958316 and rs16553624) have a MAF < 5%, and none showed differences when comparing patients against controls in either the study by Sadovnick et al. or in our own. Sadovnick and colleagues selected a series of variants of P2RX7 and P2RX4 after excluding variants detected in unaffected individuals from the same family or not observed in at least 2 patients of the same family. However, none of the selected variants were identified in any of the participants of our study. The study of these variants in 2211 patients with MS and 880 controls did not reveal an increased risk of the disease. However, Sadovnick et al.34 identified a family displaying an association between P2RX7 and P2RX4 variants and MS: 5 of the 6 affected individuals from the third generation showed the mutations in heterozygosis, although the family also included 2 unaffected mutation carriers, one in the second generation and the other in the third generation. The variants detected in that family were P2RX4 variant rs765866317 (c.403G>A; G135S) and P2RX7 variants rs140915863 (c.614C>A; T205M) and
rs201921967 (c.1082A>G; N361S). According to the authors, these variants would constitute a haplotype, with the disease following a dominant inheritance pattern with low penetrance (some individuals with the haplotype did not have MS). A functional study revealed inhibition of P2X7 pore function and increased ATP activity.34 We used the criteria established by Sadovnick et al.34 to analyse the role of the variants identified in each family, which we consider to be a very reasonable approach. In a case-control study conducted in Spain, Oyanguren-Desez et al.32 found a higher frequency of P2RX7 variants (especially rs17525809 and rs208294) in patients with MS and suggested that these variants are associated with a gain of function.
Our study is interesting in that it analyses complete families (both affected and unaffected individuals); the only missing data is from one patient with MS who died before the study began. None of the variants identified were found to be specifically associated with the disease after comparing patients against unaffected relatives. None of the participating families showed any of the variants observed in the family studied by Sadovnick et al.,34 which may point to a geographical component in variant distribution. Three families in our study displayed an association between MS and variants rs208294, rs7958311, and rs7958316; and one of the latter 2 variants is also reported by Oyanguren-Desez et al.32 The association between MS and variant rs208294 therefore seems plausible; this variant has been associated with susceptibility to sepsis53 and febrile seizures,54 and has been found to protect against rheumatoid arthritis.55 Although this variant has a gain- of-function effect due to its role as purinergic receptor, it has not been found to be associated with increased risk of rheumatoid arthritis.56
Association between variants and risk of multiple sclerosisThe P2RX7 locus is a highly polymorphic region showing areas of linkage disequilibrium; a haplotype block has been described at base positions 1042, 1070, 1379 and 1487. The alleles in the latter 2 base positions may be associated with rs208294.45,57 Oyanguren-Desez et al.32 suggested that the association of variants forming haplotypes may have an impact on the risk of MS, either increasing or reducing it. This highly polymorphic region may present associations of variants not forming haplotypes, as a mere consequence of their high frequency, even among the general population. Other studies have proposed an association between the disease and variants of such other genes as P2RX434 or CAMKK2,46 which are located in practically the same region of the chromosome. CAMKK2 codes for a protein that creates a signalling pathway made up of kinase in response to increased intracellular calcium levels58; the gene, located on 12q24.2, contains 18 exons and has been involved in multiple functions, including neuronal differentiation.59CAMKK2 has numerous variants, which had not to date been linked to MS. Although we found an association between some CAMKK2 variants and P2RX7 variants, and observed 2 variants linked to MS in 2 families, the potential relationship between these variants and the disease requires specific analysis, which is beyond the scope of the present study. In any case, our results add to existing evidence that CAMKK2 variants may be associated with P2RX7 variants.46 However, we found no association between P2RX7 and P2RX4 variants.
We analysed the potential correlation between each variant or combination of variants and the patients’ clinical characteristics: none of the variants were found to have an impact on such variables as age at disease onset or type of MS. Strikingly, however, variant rs1718119 was found specifically in type B families and was associated with sex-related variations in frequency, a rare occurrence in this type of families.39 This suggests that this variant may have a specific role in disease pathogenesis.
Gain-of-function and loss-of-function variantsIn addition to the marked polymorphism of the P2RX7 gene, one of the most relevant aspects of the study of P2RX7 is that variants have functional consequences; these do not
result from the production of altered proteins (as occurs with many mutations following DNA transcription), but rather from the fact that alterations to the efficiency of the receptor, as prolonged activation by ATP makes receptors shift from a channel function, allowing the passage of small ions, to a pore function, allowing the passage of larger molecules.1,2Table 1, adapted from Caseley et al.,60 shows the functional correlations of the variants described in the literature. A comparison of the presence of these variants in our families and in different population databases reveals that families including individuals with familial MS showed significantly higher levels of polymorphism, suggesting that these variants increase genetic susceptibility, promoting family clustering of MS when combined with environmental factors or variants in other genes. This supports the hypothesis that MS is polygenic. Presence of variants rs7958311, rs208294, rs2230912, rs1718119, and rs2230911 modify the risk of MS. A study of HEK293 cells found rs7958311 to be a gain-of-function variant; it has been suggested that nucleotide changes at this position may cause loss of function (R270C), hypofunction (R270H), or hyperfunction (H270R)61. This variant has been found to be associated with pain tolerance62,63 and gouty arthritis.64 Variant rs208294 has been shown to increase calcium entry into the receptor in response to ATP in lymphocytes from patients with chronic lymphocytic leukaemia65 and in HEK293 cells66; it is also reported to increase protein expression.67 It has also been associated with pain intolerance,61,62 febrile seizures and sepsis,68,69 and rheumatoid arthritis,70 although some researchers refute the latter association.54,71 Despite controversy as to whether rs2230912 behaves as a gain-of-function variant, the hypothesis is plausible.72 The variant has been associated with mood and affective disorders,73–77 even in family studies78 and in patients with sepsis.73 Although rs1718119 was initially considered a loss-of-function variant, it has since been observed to cause a gain of function.79,80 Copresence of rs1718119 and CAMKK2 variant rs3817190 has been associated with mood disorders81; a correlation between rs1718119 and toxoplasmosis has also been reported.82,83 Little information is available on rs2230911, although it is suggested that the variant may cause a loss of function84; it has also been associated with osteoporosis,85,86 although some studies have found no such association.87
One relevant question is whether copresence of gain-of-function and loss-of-function variants has an impact on the risk of the disease, as described in STING-associated vasculopathy with onset in infancy, a disease caused by mutations in the TMEM173 gene. Copresence of loss-of-function mutations in these patients inhibits gain-of-function variants88; this has been confirmed in vitro by introduction of a loss-of-function variant into cells.89 However, analysis of the copresence of variants did not allow us to confirm this effect.
ConclusionsFamily studies of MS complement findings from GWAS.90–92 Our study analyses the association between variants of the P2RX7 gene, which is located at the beginning of innate immune signalling pathways and may consequently be linked to risk of MS. Our most relevant finding is that families including individuals with familial MS show a higher frequency of P2RX7 polymorphisms than does the general population, regardless of whether individuals have the disease. We were unable to determine the effect of the copresence of P2RX4 and CAMKK2 variants with P2RX7 variants, or the potential effect of the haplotypes described in the gene. Our results support the hypothesis that MS is a polygenic disease influenced by environmental factors, and suggest that familial forms involve previously unknown mechanisms of genetic predisposition that could not be detected with GWAS. The fact that P2RX7 activity can be modified suggests the possibility of pharmacological prevention of the disease in families including members with familial MS.
Author contributionsLead researchers: UGP, JMAG, JMG; study design: UGP, VP, JAMG, JMG; patient assessments: JAMG, PME, LVB, JMG; family studies: VP, LTF, PME; data coordination: UGP, VP, LTF; database: UGP, VP, JAMG, LTF, JMG; data filtering and analysis: VP, LTF, JMG, JAMG; statistical analysis: VP, UGP, JMG, JAMG; analysis of results: VP, LTF, JMG, UGP, JAMG, DDOH, BSC; figures and tables: VP, LTF, JMG; manuscript drafting: UGP, JMG, VP, JAMG, LTF.
Ethics approval and consent to participateThe study was approved by the Clinical Research Ethics Committee at Hospital Clínico San Carlos, code reference # CI 16/487-E. All participants gave written informed consent. Data were handled in observance of Spanish legislation on data protection (Organic Law 15/1999 of 13 December). The project was carried out in accordance with the principles of the Declaration of Helsinki (Recommendations Guiding Physicians in Biomedical Research Involving Human Subjects, Helsinki 1964, amended October 2013).
FundingNo funding, no data.
Conflict of interestThe authors have no conflict of interest to disclose.
We are grateful to our patients and their families for the information and DNA samples provided for the study. We also wish to thank the Spanish Society of Neurology’s Research Operations Office for their help with the translation of the manuscript.
The following is Supplementary data to this article:
