



La epilepsia benigna con puntas centro-temporales (EBPCT) es el síndrome epiléptico más frecuente de la infancia, tiene carácter edad-dependiente, elevada predisposición genética y curso benigno. El objetivo de este trabajo es describir el curso clínico y el pronóstico de 60 pacientes diagnosticados de EBPCT en nuestro centro.
Pacientes y métodosRevisión retrospectiva de los pacientes diagnosticados de EBPCT en un hospital universitario (1995-2009). Se dividieron en 2 grupos: a) pacientes que cumplían todos los criterios clásicos de EBPCT, y b) cumplían los criterios excepto uno (menos de 4 años; crisis en vigilia; alteraciones EEG no típicas).
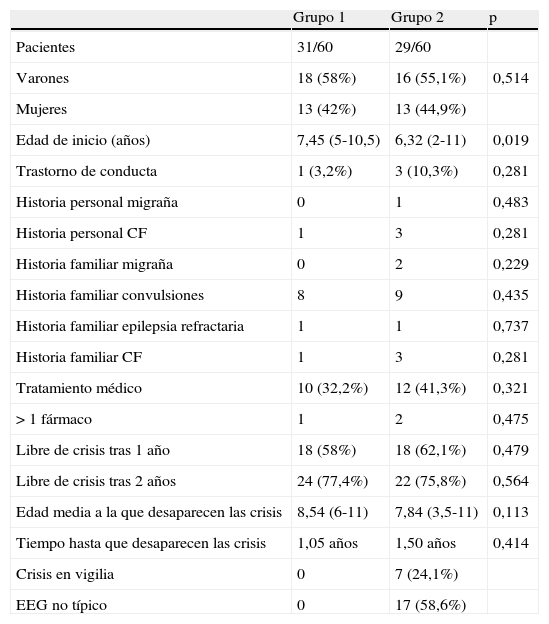
ResultadosSe seleccionó a 60 pacientes, 34 varones y 26 mujeres. Se incluyó a 31 pacientes en el grupo 1 y a 29 en el grupo 2. Edad media de inicio en el grupo 1: 7,45 años; grupo 2: 6,55 años. Se indicó tratamiento médico en 32,2% de pacientes del grupo 1, y en 41,3% del grupo 2. La evolución fue favorable en la mayoría: 58% en el grupo 1 y 62,1% en el 2 estaban libres de crisis tras un año. Edad media a la que desaparecieron: 8,54 años en el grupo 1 y 7,84 años en el grupo 2. No se encontraron diferencias estadísticamente significativas en ninguno de estos parámetros.
ConclusionesA diferencia de lo que algunos autores habían publicado, en este trabajo no se han identificado factores clínicos de mala evolución en pacientes con EBPCT, de modo que su diagnóstico se correlaciona con una evolución favorable y un excelente pronóstico neurológico.
Benign epilepsy with central-temporal spikes (BECTS) is the most common epileptic syndrome in childhood. It is an age-dependent, genetically determined and benign condition. The aim of this study is to describe the clinical course and prognosis in 60 patients with BECTS diagnosed in our hospital.
Patients and methodsWe made a retrospective review of patients diagnosed with BECTS in a University Hospital (1995-2009). They were divided into 2 groups: 1) Patients who met all BECTS classical criteria. 2) Patients who met all the criteria but one (less than 4 years; diurnal seizures; atypical EEG abnormalities).
ResultsA total of 60 patients, 34 males and 26 females were included, with 31 patients in group 1 and 29 in group 2. The mean age at onset in group 1: 7.45 years, group 2: 6.55 years. Medical treatment was indicated in 32.2% of patients in group 1 and 41.3% in group 2. The outcome was favourable in the majority: 58% in group 1 and 62.1% in group 2 were free of seizures after 1 year. Average age in which it disappeared: 8.54 years in group 1 and 7.84 years in group 2. There were no statistically significant differences in any of these parameters.
ConclusionsUnlike that published by some authors, we have not identified any poor outcome factors in patients with BECTS in this study, meaning that an accurate diagnosis correlates with a good prognosis and excellent neurological outcome.
La epilepsia benigna con puntas centro-temporales (EBPCT) se considera el síndrome epiléptico más frecuente en la infancia, alcanzando un valor porcentual del 15-24% de todos ellos1-4. La edad de inicio tiene un amplio margen (2 a 13 años), con 7 años como edad media. Es considerado un síndrome epiléptico edad dependiente con elevada predisposición genética y curso benigno1-4.
Es exigible que se presente en niños con previa normalidad neuropsicológica y curse con un fenotipo característico de epilepsia focal con presentación preferentemente en el sueño. La manifestación ictal habitual se presenta con movimientos clónicos hemifaciales, que pueden venir precedidos de síntomas somatosensoriales en labios, lengua y mejilla, asociado en ocasiones a movimientos clónicos del miembro superior e inferior ipsolaterales2-4, apareciendo durante el sueño en el 75% de los casos, habitualmente al inicio, o bien en las fases finales (despertar) de este.
Las crisis se generalizan secundariamente en el 20-54% de los casos y el EEG característico pone en evidencia puntas de alto voltaje o complejos punta-onda en la región centro-temporal, que pueden difundir al lado contralateral, todo ello con una actividad bioeléctrica cerebral de fondo normal. La neuroimagen es normal1-4.
El objetivo de este trabajo ha sido describir el curso clínico y el pronóstico a largo plazo de 60 pacientes diagnosticados de EBPCT en nuestro centro, comparando las características de los que cumplían todos los criterios clásicos con el grupo de los que los cumplían todos excepto uno.
Pacientes y métodosSe revisan retrospectivamente las historias clínicas de los pacientes diagnosticados de EBPCT en el Hospital Virgen de la Arrixaca de Murcia entre 1995 y 2009, eligiendo a 60 de todos ellos para su análisis retrospectivo, eliminando el resto por carecer de datos y/o exámenes complementarios realizados que impedían la uniformidad requerida en el estudio realizado. La evolución a epilepsia benigna atípica fue motivo de exclusión. Los casos examinados se dividen en 2 grupos. En el primero, se incluye a los que cumplían todos los criterios clásicos de EBPCT (crisis parciales nocturnas con o sin generalización secundaria; edad de inicio entre 4-13 años y desaparición antes de los 16 años; neurodesarrollo normal; EEG típico con puntas bifásicas en región/es centrotemporal/es, con actividad de fondo normal y neuroimagen normal). Este grupo queda constituido por 31 pacientes.
En el segundo grupo se incluye a los pacientes que cumplían todos los criterios clásicos de EBPCT excepto uno de los siguientes: edad de inicio antes de los 4 años; crisis predominantemente en vigilia, y alteraciones EEG no típicas de EBPCT. Este grupo se constituye con 29 pacientes.
En todos los casos examinados se recogen datos demográficos, trastornos acompañantes, historia personal y familiar, neuroimagen, evolución de las crisis y necesidad de medicación. El periodo mínimo de seguimiento tras el diagnóstico de epilepsia fue de 2 años.
Los resultados se analizan mediante el programa SPSS, utilizando la prueba de la chi al cuadrado para las variables cualitativas, como el sexo, el diagnóstico de trastorno de conducta, la presencia o no de antecedentes personales o familiares de convulsiones febriles o de migraña, la presencia de antecedentes familiares de convulsiones o epilepsia refractaria, la utilización o no de iniciar tratamiento antiepiléptico, la necesidad de emplear más de un fármaco y la ausencia de crisis tras 1 y 2 años, respectivamente.
Para comparar la edad de inicio y de finalización de la epilepsia entre ambos grupos, se utilizó la prueba de la t de Student. Para comparar el tiempo que transcurrió hasta la desaparición de las crisis se utilizó la prueba de la U de Mann-Whitney. En ambos casos se utilizó un valor de p inferior a 0,05. Los datos del análisis estadístico aparecen en la tabla 1.
Análisis comparativo de los grupos
| Grupo 1 | Grupo 2 | p | |
| Pacientes | 31/60 | 29/60 | |
| Varones | 18 (58%) | 16 (55,1%) | 0,514 |
| Mujeres | 13 (42%) | 13 (44,9%) | |
| Edad de inicio (años) | 7,45 (5-10,5) | 6,32 (2-11) | 0,019 |
| Trastorno de conducta | 1 (3,2%) | 3 (10,3%) | 0,281 |
| Historia personal migraña | 0 | 1 | 0,483 |
| Historia personal CF | 1 | 3 | 0,281 |
| Historia familiar migraña | 0 | 2 | 0,229 |
| Historia familiar convulsiones | 8 | 9 | 0,435 |
| Historia familiar epilepsia refractaria | 1 | 1 | 0,737 |
| Historia familiar CF | 1 | 3 | 0,281 |
| Tratamiento médico | 10 (32,2%) | 12 (41,3%) | 0,321 |
| > 1 fármaco | 1 | 2 | 0,475 |
| Libre de crisis tras 1 año | 18 (58%) | 18 (62,1%) | 0,479 |
| Libre de crisis tras 2 años | 24 (77,4%) | 22 (75,8%) | 0,564 |
| Edad media a la que desaparecen las crisis | 8,54 (6-11) | 7,84 (3,5-11) | 0,113 |
| Tiempo hasta que desaparecen las crisis | 1,05 años | 1,50 años | 0,414 |
| Crisis en vigilia | 0 | 7 (24,1%) | |
| EEG no típico | 0 | 17 (58,6%) |
De los 60 pacientes seleccionados, la distribución entre ambos grupos fue muy similar en número: 31 pacientes en el grupo 1 y 29 en el grupo 2, y en sexo: 34 varones y 26 mujeres. La edad media de inicio en la muestra completa fue 7,01 años (rango 2,5-11,2 años), siendo estadísticamente inferior en el grupo 2 (6,55 años) que en el 1 (7,45 años), con un valor de significación de 0,019, hallazgo no relevante y esperado, ya que uno de los criterios de inclusión en el grupo 2 fue la edad inferior a 4 años.
Las crisis se presentaron preferentemente durante el sueño o al despertar en 53 pacientes (88,3%) y en vigilia solo en 7 de ellos (11,7%), incluyéndose estos últimos en el grupo 2.
Los trastornos de conducta aparecieron en 4 pacientes (2 de ellos trastorno por déficit de atención e hiperactividad), uno de ellos en el grupo 1 y 3 en el grupo 2, diferencia sin valor estadísticamente significativo. En los pacientes con trastorno de conducta, este fue leve y mejoró con terapia cognitivo-conductual.
En 4 pacientes se refirieron antecedentes de convulsiones febriles, 3 de ellos en el grupo 2. En los antecedentes familiares únicamente se refirieron antecedentes de migraña en 2 casos, ambos del grupo 2, así como antecedentes de convulsiones en 8 pacientes del grupo 1 y en 9 del grupo 2. Tampoco estas diferencias alcanzaron valor estadísticamente significativo.
En cuanto al tratamiento, se indicó en 10 niños del grupo 1 (32,2%), y en 12 del grupo 2 (41,3%), sin que en la mayoría de los casos fuera necesario utilizar más de un fármaco (salvo un paciente del grupo 1 y 2 del grupo 2). El fármaco más utilizado fue la carbamazepina, empleada como primer fármaco en 8 pacientes, seguido del ácido valproico como primera elección en 6 de ellos. Ninguno de los pacientes revisados presentó deterioro cognitivo o empeoramiento de las crisis atribuibles a la medicación. El seguimiento a largo plazo de los pacientes tratados halla su mayor dificultad en dos aspectos: la característica retrospectiva del trabajo y el hecho de haber sido reclutados en un hospital que es referencia regional de neuropediatría hasta los 11 años, de forma que posteriormente los niños son atendidos en hospitales comarcales, habitualmente en unidades de neurología general.
Para determinar la evolución se contabilizaron el número de pacientes que estaban libres de crisis tras un año del diagnóstico y los que lo lograron tras 2 años, la edad media a la que desaparecieron las crisis y el tiempo medio que transcurrió desde el inicio hasta su desaparición. La evolución fue favorable en la mayoría de los casos, estando libre de crisis al cumplir el primer año de evolución el 58% de los del grupo 1 y el 62,1% del grupo 2. La edad media a la que desaparecieron fue algo superior en el grupo 1 (8,54 años) que en el grupo 2 (7,84 años) a pesar de que el tiempo medio que transcurrió entre el inicio y la desaparición fue superior en el grupo 2 (1,50 años) que en el grupo 1 (1,05 años).
En esta serie no se hallan diferencias significativas entre el número de pacientes que recibieron tratamiento, ni en el tiempo transcurrido hasta la desaparición de las crisis. En los 60 pacientes se realizó al menos un registro electroencefalográfico (EEG) en vigilia, completando además el estudio con un registro en sueño en 41 de ellos (68,3%).
De todo el grupo, 43 pacientes (71,6%) presentaban el registro típico, con puntas de alto voltaje o complejos punta-onda en la región centro-temporal, con o sin difusión al lado contralateral. Dentro de estos 43 niños están incluidos los 31 del grupo 1 y los 17 (58,6%) del grupo con EBPCT atípica. Los hallazgos atípicos más frecuentes fueron: puntas epilépticas adicionales en otras regiones (11 pacientes), registro normal (3 pacientes) y otros hallazgos en el EEG (otros 3 pacientes). Los pacientes con EEG normal presentaron crisis exclusivamente en sueño, en escaso número (entre 2 y 4 a lo largo de toda su evolución) no presentaron trastorno de conducta ni otras incidencias y ninguno de ellos presentó crisis más allá de los 8 años.
Se realizó estudio de neuroimagen a 42 pacientes (70%), que fue normal todos ellos, salvo uno, que presentaba como hallazgo casual una malformación Arnold-Chiari tipo I, obviamente sin relación fisiopatogénica con las crisis epilépticas. Los datos demográficos, la evolución clínica y la comparación entre ambos grupos se reflejan en la tabla 1.
DiscusiónLos hallazgos atípicos en la EBPCT pueden establecerse en función del tipo de crisis (crisis exclusivamente diurnas, parálisis post-ictal, crisis prolongadas) o de las características del EEG (morfología atípica de las puntas, localización inusual, descargas punta-onda o actividad de fondo anómala), aunque la edad precoz de inicio (inferior a 4 años) parece ser el factor más determinante en la progresión atípica del cuadro5-8, que algunos autores han relacionado con mayor probabilidad de dificultades de aprendizaje y trastorno de conducta9.
Las características clínicas y del EEG de los niños incluidos en este trabajo coinciden con la literatura previa, con una edad de inicio entre 2,5 y 11,2 años.
La evolución neuropsicológica en los niños con EBPCT es normal y los trastornos de conducta son menos frecuentes que en otros síndromes epilépticos pediátricos1-4. Sin embargo, en los últimos años se han publicado trabajos que describen en estos pacientes una mayor probabilidad de trastorno de lenguaje y lecto-escritura, dificultades de aprendizaje y/o déficit de atención9-12.
De hecho, algunos autores4,6,13 sostienen que la EBPCT y sus diferentes formas atípicas (incluidos la epilepsia con punta-onda continua durante el sueño lento y el síndrome de Landau-Kleffner) forman parte de un espectro con la misma base genética, pero con diferencias en la duración, la localización y la difusión de la actividad epiléptica.
Algunas de las formas atípicas pueden ser diferentes formas evolutivas en el mismo paciente, desencadenadas por algunos fármacos antiepilépticos4,6,13.
En el presente trabajo, al igual que en otros previos, se ha observado una elevada incidencia (50%) de pacientes que no cumplían todos los criterios clásicos de EBPCT, hallazgo relacionado con los criterios empleados para definir las variantes clínicas, que difiere entre los distintos autores. Así, Verrotti et al.9 incluyen en el grupo «atípico» a los pacientes con otros tipos de crisis; Tavares et al.8 incluyen a los pacientes con otros tipos de crisis, pacientes con crisis exclusivamente en vigilia y alteraciones atípicas en el EEG; Datta y Sinclair5 y Callenbach et al.7 incluyeron, además, a pacientes menores de 4 años con dificultades de aprendizaje, anomalías en el examen neurológico, otros tipos de crisis y/o anomalías en el EEG atípicas. Nosotros hemos empleado estos últimos criterios de inclusión.
Algunos de los trabajos mencionados5,7-9 han tratado de identificar factores predictivos de pronóstico, pero ninguno de ellos se relaciona claramente con peor evolución neuropsicológica ni con mayor duración de la epilepsia. En la serie de Datta y Sinclair5, el grupo atípico presentó crisis más difíciles de controlar, aunque idéntico pronóstico. En la de Verrotti et al.9, el grupo atípico presentó más problemas de aprendizaje y de conducta, aunque estos resultados no han sido refrendados por otros autores7,8. En nuestro trabajo, en el grupo 2 se apreció una mayor proporción de pacientes con trastorno de conducta, pero sin valor estadísticamente significativo. Será necesario realizar estudios más amplios y prospectivos que permitan corroborar o desmentir este resultado. Teniendo en cuenta el excelente pronóstico que presentan estos pacientes en general, resulta complicado identificar a los que no lo presentarán, sin que tampoco los resultados obtenidos en nuestro estudio permitan establecer un grupo de pacientes con EBPCT a los que se les pueda atribuir a priori un peor pronóstico, ya que la evolución de la epilepsia es muy similar en ambos grupos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr., Juan Ignacio Ortuño Sempere, pediatra del Hospital Los Arcos del Mar Menor, cuya colaboración en el estudio estadístico ha sido fundamental.

