




Focal tumour-like demyelinating lesions are defined as solitary demyelinating lesions with a diameter greater than 2cm. In imaging studies, these lesions may mimic a neoplasm or brain abscess; as a result, invasive diagnostic and therapeutic measures may be performed that will in some cases increase morbidity. Our aim was to analyse and characterise these lesions according to their clinical, radiological, and pathological characteristics, and these data in addition to our literature review will contribute to a better understanding of these lesions.
MethodsThis descriptive study includes five cases with pathological diagnoses. We provide subject characteristics gathered through reviewing their clinical, radiology, and pathology reports.
ResultsPatients’ ages ranged from 12 to 60 years; three patients were female. The time delay between symptom onset and hospital admission was 3-120 days. Clinical manifestations were diverse and dependent on the location of the lesion, pyramidal signs were found in 80% of patients, there were no clinical or radiological signs of spinal cord involvement, and follow-up times ranged from 1 to 15 years.
ConclusionBrain biopsy is the gold standard for the diagnosis of demyelinating tumour-like lesions; however, their clinical features, along with several magnetic resonance imaging features such as open ring enhancement, venular enhancement, the presence of glutamate in spectroscopy, and others may be sufficient to differentiate neoplastic lesions from focal tumour-like demyelinating lesions.
Las lesiones desmielinizantes focales seudotumorales se definen como lesiones solitarias, desmielinizantes, con diámetro superior de 2cm. Estas pueden imitar mediante el estudio imagenológico una neoplasia o absceso cerebral, lo que lleva a medidas diagnósticas y terapéuticas invasivas en algunos casos, incrementando la morbilidad. Nuestro objetivo fue analizar y caracterizar estas lesiones clínica, radiológica y patológicamente, lo que, sumado a la revisión de la literatura, aportará al entendimiento de este tipo de trastornos.
MétodosEn este estudio descriptivo, se reportan 5 casos con diagnóstico patológico. Mediante la revisión de informes relacionados, clínicos, radiológicos y patológicos, se resumen las características de los sujetos.
ResultadosLa edad de los pacientes osciló entre los 12 y los 60 años, 3 pacientes fueron de género femenino. La latencia de los síntomas hasta admisión hospitalaria fue entre 3 y 120 días, las manifestaciones clínicas fueron diversas y dependientes de la localización de la lesión, en el 80% de los pacientes se encontraron signos piramidales y no se encontraron clínica o imagenológicamente lesiones de la médula espinal; el seguimiento de los pacientes abarca desde un año hasta 15 años.
ConclusiónLa biopsia cerebral es el estándar de oro para el diagnóstico de las lesiones desmielinizantes seudotumorales; no obstante, las características clínicas, junto con varias características de la resonancia magnética, tales como el realce en anillo abierto, el realce venular y la presencia de glutamato en la espectroscopia, entre otras, pueden ser satisfactorias en la diferenciación de las lesiones desmielinizantes focales seudotumorales de lesiones neoplásicas.
Primary demyelinating disorders of the central nervous system (CNS) are a set of entities that include acute disseminated encephalomyelitis (ADEM), acute haemorrhagic leukoencephalopathy, neuromyelitis optica (Devic disease), and several types of multiple sclerosis (MS). The latter includes chronic MS (Charcot type), acute MS (Marburg type), myelinoclastic diffuse sclerosis (Schilder type), and concentric sclerosis (Baló type). Charcot or classic MS is the most frequent and best-known variant, and although its prevalence is low in tropical regions,1,2 it is currently on the rise. However, there is a rare demyelinating disease that presents as a large area of focal demyelination (>2cm) associated with a mass effect which can imitate a tumour or cerebral abscess in an imaging study. The literature refers to this disease as ‘demyelinating pseudotumour’, ‘tumour-like demyelinating lesions’, and ‘swollen demyelinating lesions’.
Although magnetic resonance imaging (MRI) has good sensitivity to detect these lesions, it is not a specific test for demyelinating pseudotumour. A neuroradiological study showing a large lesion located in the deep white matter with a significant mass effect, which clinically manifests as signs of increased intracranial pressure, is very indicative of a CNS tumour. In rare cases, a demyelinating disease can manifest with atypical symptoms and images that indicate a brain tumour. These focal tumour-like demyelinating lesions (FTDL) may pose diagnostic challenges for both doctors and radiologists.
The difficulty involved in diagnosing a focal tumour-like demyelinating lesion often leads to a surgical biopsy, which is currently the gold standard for a definitive diagnosis.
We report five cases in which diagnosis was confirmed by a pathology study. After reviewing related clinical, radiological, and pathological reports, we summarise their diagnostic characteristics.
MethodsThis descriptive study presents five cases with a diagnosis confirmed by pathology study. We reviewed patient clinical histories to extract clinical data such as presentation profile, clinical manifestations, treatment applied, and response to treatment. Patients’ clinical outcomes were also recorded. Characteristics of the lesions on the magnetic resonance images were reviewed, and we describe microscopic findings from the lesions where possible.
ResultsAge at which lesions appeared ranged from 12 to 60 years; 3 of the 5 patients were women. Time from symptom onset to hospital admission ranged from 3 days to 4 months. Clinical manifestations varied greatly and were dependent on lesion location; however, 80% of the patients presented pyramidal signs. No spinal cord lesions were detected by clinical signs or neuroimaging tests in any of the cases. Follow-up periods ranged from one year to 15 years (Table 1).
Main clinical and paraclinical characteristics of patients
| Case | Age | Sex | Findings at symptom onset | Imaging findings | Pathology findings | Treatment | Follow-up |
|---|---|---|---|---|---|---|---|
| 1 | 39 | M | Headache, vertigo | Right frontal and temporal lesions, with central hyperintensity, cyst-like appearance, gadolinium-enhanced rim, lesions on basal ganglia, periventricular lesions, lesions on corpus callosum, and optic radiation. | H&E stain shows a blood vessel surrounded by leukocytes, which are in turn ringed by foamy macrophages.Small benign gemistocytic astrocytes.Bielschowsky silver stain shows areas of myelin loss and a significant number of preserved axons. | Alferon® over 10 years, followed by beta 1A interferon. | 15 years of follow-up with no relapses. |
| 2 | 12 | F | Acute vision loss in RE with no treatment; aphasia and right hemiparesis 4 months later. | Right frontal lesion with hyperintensity on T2-weighted image, hyperintensity on T1-weighted image, and adjacent vasogenic oedema and slight mass effect on FLAIR image. Images indicative of vessel-like structures in lesion interior. Brain perfusion shows hypoperfusion and spectroscopy with an elevated choline peak at 3.23ppm, a low NAA peak at 2.05ppm, and glutamate and glutamine peak of 2.4-2.5ppm. | LFB and H&E stains. Abundant foamy macrophages were observed in absence of coagulative necrosis, atypical reactive astrocytes (Creutzfeldt-Peters cells). Venules with perivascular lymphocytic cuffing. LFB stain shows areas of demyelination with clearly defined edges and a tendency to converge. | Short-term treatment with methylprednisolone was administered for 5 days, followed by 5 cycles of plasmapheresis. No immunomodulatory treatment was initiated. | Total recovery of strength on the right side of the body and ability to speak; RE optic atrophy remained.At one year of follow-up, no relapses had been reported. |
| 3 | 23 | F | One month before symptom onset, patient presented homonymous hemianopsia with paraesthesia of all 4 limbs. The patient was asymptomatic at the time of diagnosis. | Lesion in occipital white-matter that is hyperintense in T2-weighted sequence and hypointense in the T1-weighted and FLAIR sequences. Brain perfusion shows hypoperfusion and spectroscopy with elevated choline peak at 3.23ppm, low NAA peak at 2.05ppm, and glutamate and glutamine peak of 2.4-2.5ppm. | No biopsy required | No treatment administered. | 6 years later, physical examination revealed Babinski sign; MRI scan revealed new lesions which called for starting treatment with interferon. |
| 4 | 60 | F | Syncopal episodes and left-sided hemiparesis evolved over one month. | Cranial CT revealed right hypodense cortical-subcortical lesion indicative of glioblastoma; cerebral biopsy was performed. | H&E, PAS, and tolonium chloride staining revealed a lesion with an extensive demyelinating component, reactive astrocytosis, and vessels with thickened walls and reduced lumen. Myelin is phagocytosed by microglia; lymphocytic infiltrates in the vessel walls. | Unknown, the patient did not remain in follow-up. | Unknown |
| 5 | 42 | M | Upper left limb paresis progressing over 5 days. | Rounded lesion on the postcentral gyrus cortex on the right hemisphere, hypointense in T1-weighted sequence and hyperintense in T2-weighted sequence. No significant oedema was observed. Lesion displayed gadolinium uptake in a rim enhancement pattern with regular edges. | Focal demyelinating process with abundant macrophages, mainly surrounding blood vessels; preserved axons, discrete inflammatory lymphocytic infiltrates, and abundant reactive astrocytes. | Acute treatment with methylprednisolone over 5 days. | Complete recovery; follow-up for 8 years with no relapses. |
F, female patient; H&E, haematoxylin and eosin; LFB, Luxol Fast Blue; M, male patient; NAA, N-acetyl aspartate; RE, right eye; MRI, magnetic resonance imaging; PAS, Periodic acid-Schiff.
Demyelinating lesions resembling cerebral tumours have been described since the 1970s and 1980s, and differential diagnosis in these cases required a brain biopsy.3,4 In 1993, Kepes reported on 31 patients, 24 with solitary lesions and seven with multifocal lesions.5 These lesions resembled tumours, abscesses, neurocysticercosis cysts, gliomas, or metastasis. All patients underwent cerebral biopsy. This type of demyelinating disease was called tumour-like or swollen demyelination because of its appearance on MRI images. Kepes initially interpreted these lesions as an intermediate disorder between MS and ADEM. Some authors now classify them as MS variants, since clinical symptoms and radiological findings of Charcot MS appear in some cases. Lesions in other cases, however, are isolated, follow a monophasic course, and resolve completely. As a result, experts are not in full agreement as to the nature of the disease. The 31 patients included in the study by Kepes5 can be considered ‘index cases’, and we therefore propose calling them ‘Kepes FTDL’, in honour of this leading neuropathologist who died in February 2010.6 Lucchinetti et al.7 subsequently described the largest series of which we are aware: 168 patients with histopathologically confirmed demyelinating lesions larger than 2cm.
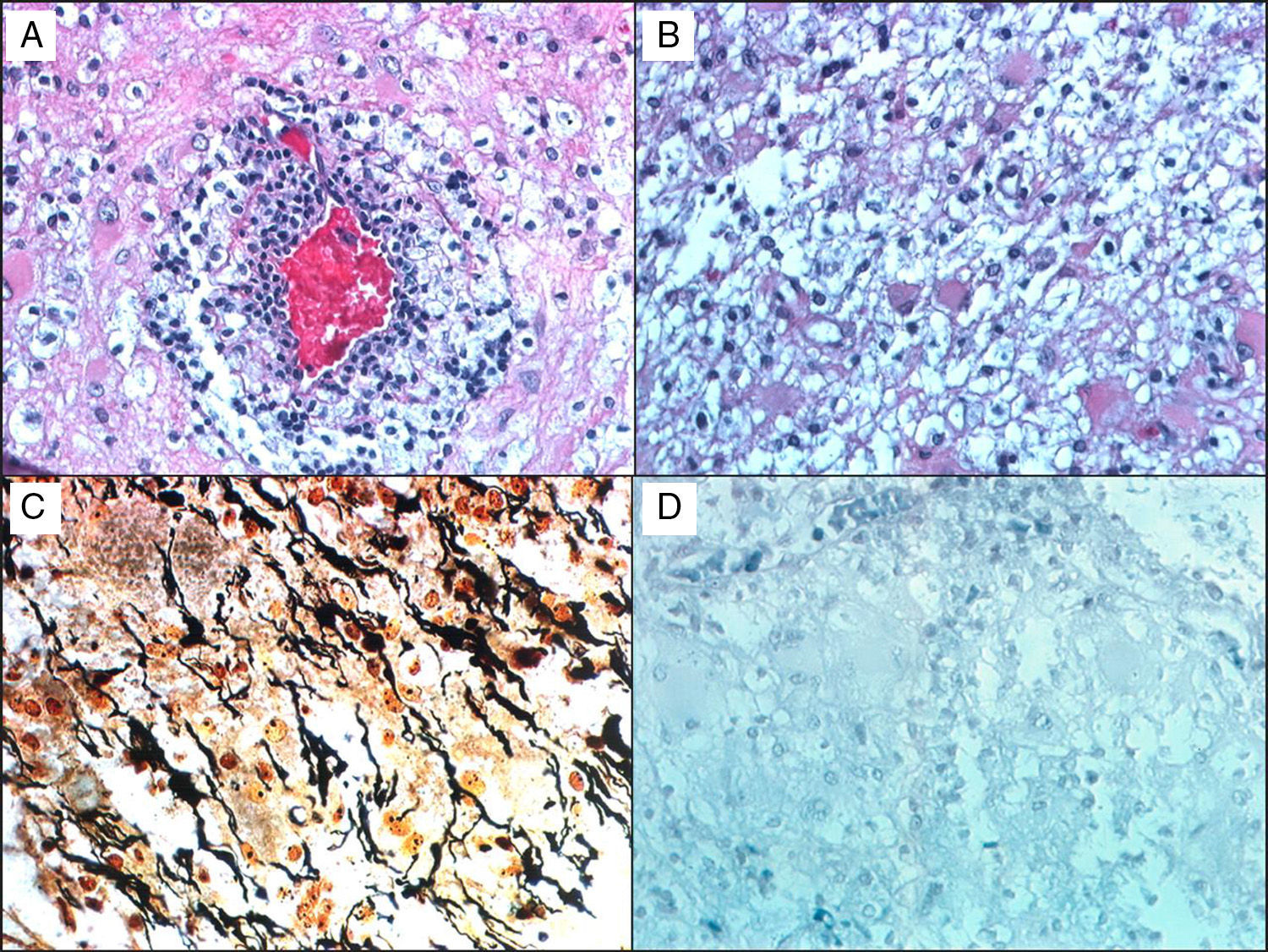
Histological studies of these lesions showed chronic perivascular inflammatory infiltrates, macrophages containing myelin degradation products, reactive astrocytes, demyelinating lesions with clearly defined margins, and relative axon preservation (Fig. 1). An acute demyelinating process can mimic a malignant glioma, which can contain pleomorphic astrocytes including Creutzfeldt cells. The pathological patterns of demyelinating disorders include features that made them difficult to distinguish from gliomas,8 such as pleomorphic astrocytes, hypercellularity, necrosis, mitotic figures, and cyst-like changes. These pleomorphic astrocytes, including multinucleated or Creutzfeldt cells, may be highly noticeable in active plaques, especially near the edge. They tend to display a homogeneous distribution pattern opposite to that seen in gliomas, in which cells tend to agglomerate. In contrast with neoplastic astrocytes, reactive astrocytes are not characterised by a hyperchromatic nucleus. Meanwhile, macrophages may be mistaken for astrocytes or oligodendroglia and are also indicative of infiltrative tumour growth. Diagnosis therefore requires immunohistochemical stains that affect macrophages (anti-HAM 56 or CD68, for example), together with myelin staining to evidence the demyelinating process (Fig. 1).
 and Dr Toro (Bogotá): (a) H&E stain showing a vessel surrounded by leukocytes, surrounded in turn by foamy macrophages. (b) Small benign gemistocytic astrocytes. (c) Bielschowsky silver stain that shows areas of myelin loss and a significant number of preserved axons. (d) Weil and Weigert stain showing almost total myelin loss. Findings were compatible with demyelinating disease. The patient was monitored for 15 years, and displayed an excellent response to interferon treatment.")
A 38-year-old man with symptoms of headache and vertigo. MRI revealed multiple right frontal and temporal lesions, lesions on basal ganglia, periventricular lesions, lesions on corpus callosum, and optic radiation. Central hyperintensity with a cyst-like appearance and gadolinium-enhanced rim was observed on T2-weighted images. Results from the neurological examination were normal. Biopsy was evaluated by Dr Kepes (Kansas) and Dr Toro (Bogotá): (a) H&E stain showing a vessel surrounded by leukocytes, surrounded in turn by foamy macrophages. (b) Small benign gemistocytic astrocytes. (c) Bielschowsky silver stain that shows areas of myelin loss and a significant number of preserved axons. (d) Weil and Weigert stain showing almost total myelin loss. Findings were compatible with demyelinating disease. The patient was monitored for 15 years, and displayed an excellent response to interferon treatment.
Biological and clinical findings from these patients differ from those of patients with acute fulminant multiple sclerosis (Marburg type)9 and Schilder disease.10–12 The report describes clear differences in the course of the disease. Most patients in the Kepes et al. series presented a monophasic course, but lesions may also be recurrent.5 The clinical course of monophasic lesions may be caused by the relative axonal integrity present here, in contrast with the pronounced axonal loss that occurs in chronic MS. In the series by Lucchinetti et al. 70% of the patients developed recurrent lesions.7 Definitive diagnosis of MS according to McDonald criteria was assigned after a median follow-up of 3.9 years, and median time to second exacerbation of 4.8 years.
Diagnosis of this type of demyelinating lesion initially required a brain biopsy but thanks to the advances in CT and MRI techniques, biopsies are becoming less frequent. We now describe the neuroimaging sequences that are useful for diagnosing this type of lesions.
Simple CT is useful in differential diagnosis of gliomas when compared to MRI results, since the rim observed in contrast-enhanced T1-weighted sequences is isointense, unlike the hypodense ring observed on CT in demyelinating lesions. This rim is sometimes not discernible by CT imaging but appears in T2 and contrast-enhanced T1 MR images.13 Contrast-enhanced CT sometimes shows an incomplete rim similar to that observed using MRI, as described below.14
Conventional MRI sequences show large single or multifocal lesions usually evidencing no mass effect, no cortical involvement, and little oedema. Oedema, together with incomplete rim on gadolinium-enhanced T1-weighted MRI sequences, has a P value of <.05.13 Contrast-enhanced areas indicate active demyelinating lesions affecting the white matter. In the case of tumour-like lesions or abscesses, the rim is generally complete.15 Detecting other typical MS demyelinating lesions in the brain or cervical spinal cord may also aid differential diagnosis.14 Another technique is dynamic T2*-weighted gradient-echo imaging, which is performed during IV administration of the contrast agent. These images show vessel-like structures running from the centre of the lesion towards a subependymal vein that is usually dilated.16 The diagnostic accuracy of CT plus MR imaging is 97%, compared to 73.0% for MR imaging alone (P=.001).13
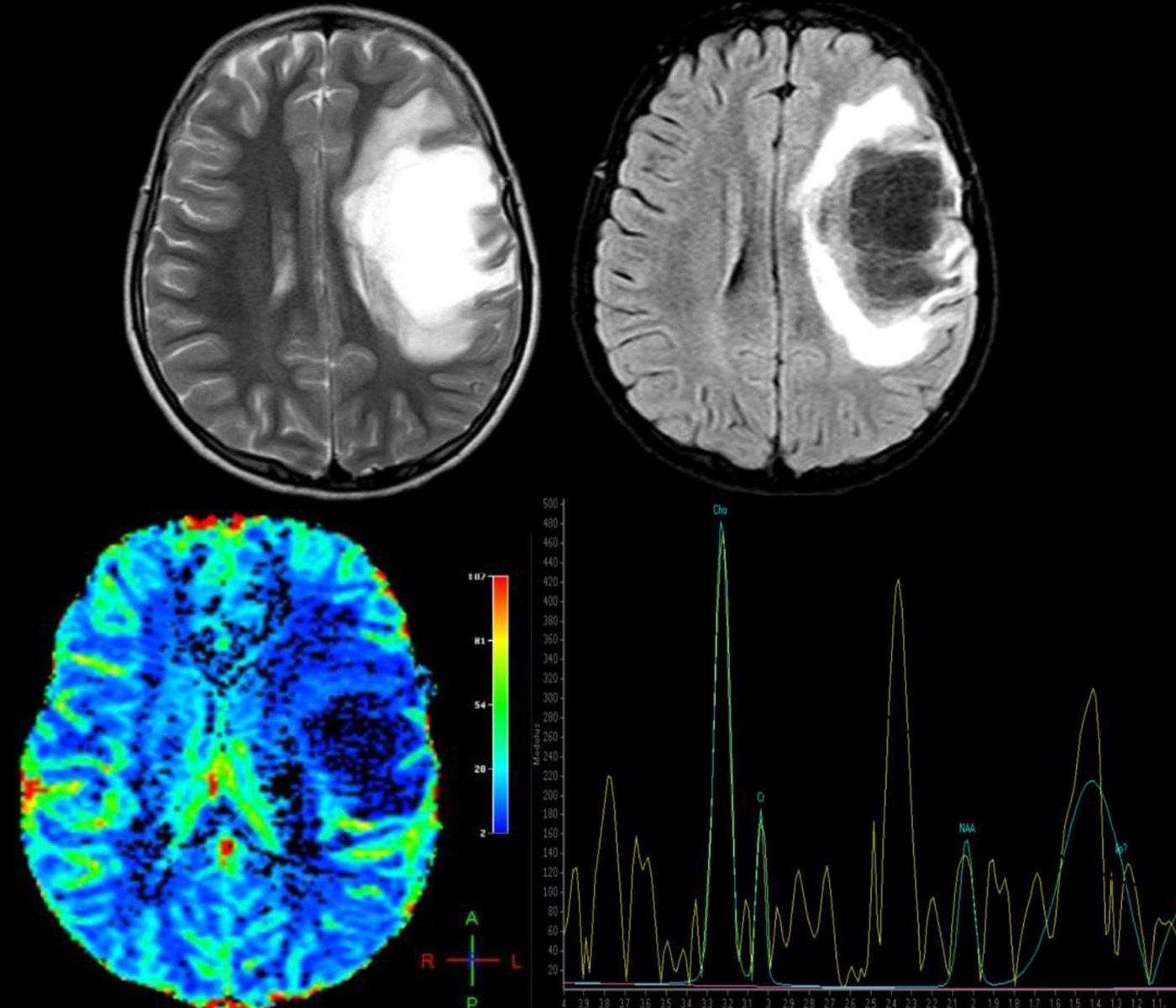
Advanced MRI techniques are equally useful in the differential diagnosis of these lesions. MR spectroscopy reveals a depressed peak for N-acetyl aspartate (NAA), which indicates neuronal death. There are also elevated choline (CHO) and lipid (LIP) peaks, which are consistent with cell membrane rupture; and elevated lactate (LAC), probably due to activated macrophages or ischaemia secondary to an acute inflammatory process.17,18 Nevertheless, these findings are highly non-specific and their validity is therefore limited, leading some authors to conclude that spectroscopy cannot be used to distinguish between neoplasm and demyelinating lesions.19 However, once the demyelinating process becomes chronic, CHO, LIP, and LAC peaks normalise and the NAA peak remains persistently low. On the contrary, in cases of neoplasm, metabolites remain altered; some authors recommend starting anti-inflammatory treatment and performing follow-up spectroscopy to distinguish between the 2 processes when the diagnosis is unclear.17 Cianfoni et al.20 also reported an interesting finding in short echo time MR spectroscopy. They observed marked elevation of glutamate and glutamine peaks, which is not typically seen in intra-axial neoplastic processes; this was thought to be secondary to an astrocyte reaction (Fig. 2).
 that was not treated. Four months later, she experienced aphasia and right hemiparesis. This figure shows some of the brain MR images that were ordered: above, axial T2-weighted images and axial Fluid Attenuation Inversion Recovery (FLAIR) images. We observe a right frontal lesion that is hyperintense in T2-weighted images and hypointense in T1-weighted images. FLAIR sequence shows adjacent vasogenic oedema and slight mass effect on the midline and ventricular system. Images suggesting vessel-like structures were obtained from the inside of the lesion. Below: brain perfusion image showing decreased enhancement and MRI spectroscopy that shows an elevated choline peak at 3.23ppm (indicative of gliolysis and remyelination processes), a decreased NAA peak at 2.05ppm (indicative of neuronal/axonal injury), and glutamate and glutamine peak of 2.4–2.5ppm. These findings were compatible with tumour-like demyelinating disease. Histopathological findings from the brain biopsy were consistent with FTDL. Doctors started a 5-day course of treatment with methylprednisolone, followed by one cycle of plasmapheresis. Clinical response was excellent. The patient completely regained her strength on the right side of the body and her ability to speak, but optic atrophy of the RE remained. After one year of follow-up, no relapses have been reported.")
A 12-year-old girl with no relevant medical history presented acute vision loss in the right eye (RE) that was not treated. Four months later, she experienced aphasia and right hemiparesis. This figure shows some of the brain MR images that were ordered: above, axial T2-weighted images and axial Fluid Attenuation Inversion Recovery (FLAIR) images. We observe a right frontal lesion that is hyperintense in T2-weighted images and hypointense in T1-weighted images. FLAIR sequence shows adjacent vasogenic oedema and slight mass effect on the midline and ventricular system. Images suggesting vessel-like structures were obtained from the inside of the lesion. Below: brain perfusion image showing decreased enhancement and MRI spectroscopy that shows an elevated choline peak at 3.23ppm (indicative of gliolysis and remyelination processes), a decreased NAA peak at 2.05ppm (indicative of neuronal/axonal injury), and glutamate and glutamine peak of 2.4–2.5ppm. These findings were compatible with tumour-like demyelinating disease. Histopathological findings from the brain biopsy were consistent with FTDL. Doctors started a 5-day course of treatment with methylprednisolone, followed by one cycle of plasmapheresis. Clinical response was excellent. The patient completely regained her strength on the right side of the body and her ability to speak, but optic atrophy of the RE remained. After one year of follow-up, no relapses have been reported.
Perfusion MRI techniques are also very useful in differential diagnosis of neoplastic and demyelinating processes. In neoplastic processes (both gliomas and CNS lymphomas), regional cerebral blood volume (rCBV) is markedly elevated and mean transit time (MTT) is decreased due to angiogenesis. In demyelinating lesions, however, rCBV is not elevated and may in fact be low, whereas MTT will be elevated (Fig. 2).14,16
A case of tumefactive demyelinating lesion due to tacrolimus neurotoxicity was recently reported.21 The patient had received a liver transplant for hepatocellular carcinoma. The potential mechanism for demyelination is mediated by immunity due to a supposed increase in T-cell activation. There have been numerous cases of other types of demyelination without a defined relationship, including a case of optic neuritis triggered by a Plasmodium falciparum infection.22 These reports illustrate the lack of knowledge of these diseases and the variability of probable factors eliciting demyelination.
We should stress that no results have a level of evidence sufficient for drawing up management guidelines, considering the low prevalence rates of these types of demyelination compared to MS.
There are both similarities and differences between treatment for MS and for these acute focal demyelinating lesions. As first-line treatment, an IV bolus of methylprednisolone dosed at 1g/day for 3 to 5 days should be administered. Episodes of acute CNS demyelination are considered refractory to corticosteroids if no improvement of at least one point on the extended disability status scale is observed in the 7 days after finishing steroid treatment.23 Weinshenker et al.24 propose the following criteria to start plasmapheresis: minimum duration of deficit 21 days from onset and 14 days from the onset of IV steroid therapy, or 12 days from onset if the deficit continues to worsen beyond 5 full days after starting steroid treatment. If any one of these criteria is met, plasmapheresis should be considered as second-line treatment. Some particularities of this type of therapy should be made clear. The proposed action mechanism of plasmapheresis for these diseases involves extraction of plasma and humoral factors in pathogenesis. This means that plasmapheresis could be beneficial in treating disorders mediated by the humoral system, such as ADEM, neuromyelitis optica,25,26 and MS with demyelinating pattern II as proposed by Lucchinetti27 (characterised by immunoglobulin and activated complement deposits). A small clinical trial of this therapy in 10 patients with pattern II found moderate to marked improvement;28 some authors suggest that the same type of damage is observed in Marburg type MS.29,30 Baló disease, which follows demyelination pattern III27 and whose pathogenesis does not involve humoral factors, was also treated in that clinical trial. No response to plasmapheresis treatment was reported in the six patients with that pattern.28 Three studies evaluate predictors of good response to treatment in demyelinating disorders.26,31,32 The most commonly listed factors with statistical validity are early treatment, short disease duration, preserved deep tendon reflexes, retrospective diagnosis of MS, and rim-enhancing lesion on MRI.
The next step is less clear since there have been only anecdotal reports of cases refractory to steroid treatment and plasmapheresis in which long-term treatments including interferons, cyclophosphamide,33–36 and mitoxantrone37 were administered, together with support treatment for complications typical of this severe and extensive involvement, such as intracranial hypertension.
ConclusionsEfforts should be directed urgently towards increasing our knowledge of FTDL, especially because of their different behaviours compared to other demyelinating disorders.
In general, monophasic tumour-like lesions have an acceptable or even excellent prognosis in some cases, unlike recurrent lesions and aggressive and even lethal types including Marburg type MS.
In most cases, diagnosis is reached after examining symptoms and neuroimaging findings. The latter are extremely significant, especially contrast-enhanced incomplete rim, peripheral diffusion restriction, contrast-enhanced veins, presence of glutamate in MR spectroscopy, and absence of increased cerebral blood flow in perfusion MR images. However, an unequivocal diagnosis requires brain biopsy.
No results present a level of evidence sufficient to issue management guidelines for these less well-known myelin disorders. Therefore, treatment for these types of demyelination resembles those administered for classic MS during relapses. Steroids at high doses are used as first-line treatment; if there is no response, plasmapheresis is performed. In some refractory cases, immunosuppressants such as cyclophosphamide and mitoxantrone have been used.
Conflict of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Jiménez Arango JA, Uribe Uribe CS, Toro González G. Lesiones desmielinizantes focales seudotumorales. Neurología. 2015;30:97–105.
