



Despite advances in the understanding of genetic and molecular aspects of Amyotrophic Lateral Sclerosis (ALS), a rapidly progressive and fatal neurodegenerative disease, the exact pathogenic mechanisms are still largely unknown. For over 20 years, numerous in vitro and in vivo studies have demonstrated the existence of a complex interaction between motor neurons and astrocytes in neurodegeneration. In ALS, astrocytes acquire a reactive phenotype through a phenomenon known as astrogliosis, in which they lose their normal functions and/or acquire highly damaging functions, altering the function and survival of motor neurons. For this review, we set out to analyse the role of astrocytes, particularly in the study of mutations in the SOD1, C9orf72, and TARDBP genes, which are closely related to the pathogenesis of familial ALS. The observations made in this study strongly suggest that the role of astrocytes in ALS is multidimensional, and specifically that astrocytes with different genetic mutations linked to ALS present diverse underlying molecular patterns. Therefore, these cells constitute an extremely promising therapeutic target in the treatment of this neurodegenerative disease.
Pese a los avances en la comprensión de las bases genéticas y moleculares de la Esclerosis Lateral Amiotrófica (ELA), una enfermedad neurodegenerativa rápidamente progresiva y mortal, aún se desconocen casi por completo los mecanismos exactos de este padecimiento. Por más de 20 años, diversos estudios in vitro e in vivo han demostrado la existencia de una interacción compleja entre las motoneuronas y los astrocitos en el evento de neurodegeneración. En la ELA, los astrocitos adoptan un fenotipo reactivo conocido como astrogliosis, en el cual pierden sus funciones normales y/o adquieren funciones sumamente dañinas, alterando la función y la supervivencia de las neuronas motoras. Para la presente revisión, nos propusimos analizar el papel que desempeñan los astrocitos, particularmente en el estudio de los genes mutantes SOD1, C9ORF72 Y TARDBP, los cuales están estrechamente relacionados con la patogenia de la ELA familiar. Las observaciones realizadas en este trabajo sugieren fuertemente que los astrocitos tienen funciones multidimensionales en la ELA, y específicamente los astrocitos con diferentes mutaciones genéticas ligadas a la ELA presentan diversos patrones moleculares subyacentes. Por lo tanto, estas células se muestran como objetivos terapéuticos sumamente prometedores en el tratamiento de esta enfermedad neurodegenerativa.
Amyotrophic Lateral Sclerosis (ALS) is a multisystem neurodegenerative disorder characterised by neuropathological, clinical, and genetic heterogeneity. It presents with rapidly progressive degeneration of both upper (projecting from the cerebral cortex to the spinal cord) and lower motor neurons (projecting from the brainstem to different muscle groups), resulting in atrophy of the muscles involved in breathing, swallowing, speech, and limb movement.1
In the United States and Europe, the incidence of ALS is estimated at 1–2 cases/100 000 person-years, with a prevalence rate of 10–12 cases/100 000 population.2 In 2018, the total number of cases in Mexico was estimated at 6000.3 Both incidence and prevalence increase with age; men are twice as likely to be affected as women, specifically in non-genetic forms of ALS. However, genetic forms are more likely to manifest in late adolescence or in early adulthood.2
Symptoms vary between patients, and disease onset may be spinal or bulbar. In both forms, progressive paralysis leads to the death of the patient due to neuromuscular respiratory failure; life expectancy after symptom onset is typically 2–3 years.4
According to etiology, ALS can be classified as sporadic (non-hereditary) or familial (hereditary). Sporadic ALS accounts for 90% of cases, and may be caused by an interaction between genetic and environmental factors; in other words, environmental factors can trigger the disease, whereas genetic mutations increase the likelihood of developing it. The remaining 10% of cases of ALS are familial, with the majority following an autosomal dominant inheritance pattern.5
Both in sporadic and in familial forms, disease pathology involves mutations in one or more genes related to ALS. These can be divided into causal genes, which cause ALS when they present a mutation, and susceptibility genes, which increase the likelihood of developing the disease. Causal genes include SOD1 (superoxide dismutase 1), C9orf72 (chromosome 9 open reading frame 72), and TARDBP (transactive response [TAR] DNA-binding protein 43 [TDP-43]). Examples of susceptibility genes are SMN1 (survival of motor neuron 1), SMN2 (survival of motor neuron 2), and ANG (angiogenin).6–10
SOD1 was the first gene associated with ALS,6 in 1993; since then, over 187 genetic variants have been shown to be involved in the risk of developing the disease, and over 30 genes have been linked to familial, sporadic, or both forms of ALS.7–10
A growing body of evidence has shown that ALS does not exclusively affect neurons; rather, it also involves and requires other cell types, including macrophages, microglia, oligodendrocytes, and astrocytes.11–14 It should be noted that astrocytes, specifically, play a role in the cascade of events that eventually leads to motor neuron death, especially in ALS associated with mutations in SOD1, C9orf72, and TARDBP. They are also involved in the neurotoxicity mechanism resulting from alterations in glutamate uptake. Both mechanisms are the result of mutations in one or more of these genes.15
Furthermore, in the adult nervous system, neural stem cells can differentiate into cells of all neural phenotypes (neurons, astrocytes, and oligodendrocytes), in accordance with the molecular signals received from their environment. The presence of mutations altering the genetics of astrocyte-lineage neural stem cells can cause anomalies in their differentiation to any neural lineage,2 leading to loss of normal homeostatic functions in the nervous system and increasing the likelihood of developing neurological disease.
Interaction between astrocytes and motor neuronsAstrocytes play a crucial role in CNS homeostasis. These cells originate in the neuroepithelium and share the same progenitor (epithelial cells) as neurons and oligodendrocytes.16,17 Astrocytes, together with the microglia, provide metabolic and trophic support to neurons and are responsible for synaptic pruning, ionic and neurotransmitter equilibrium, etc.18,19
With respect to the interaction between astrocytes and motor neurons, an astrocyte can be in contact with 4–10 neurons, hundreds of dendrites, and thousands of synapses. Astrocytes are the main element in the microenvironment of motor neurons, and any alteration affecting them will have repercussions for the nearby neurons.5
Alterations to CNS homeostasis in ALS result in the appearance of reactive or activated astrocytes (toxic phenotypes), giving rise to the phenomenon of astrogliosis, in which glial cells lose vital functions, degenerate, and contribute significantly to the death of motor neurons by secreting neurotoxic factors.20 Astrogliosis at clinical onset of ALS and neuron death have been observed in different experimental models.21,22
No significant phenotypic differences have been reported between the familial and sporadic forms of ALS. In vivo and in vitro studies report activated microglia, astrogliosis, neurons and astrocytes with protein deposits (TDP-43 inclusions, ubiquilin 2 aggregates, and intracytoplasmic SOD1 protein deposits), and apoptotic cell death.5 TDP-43 inclusions are toxic in themselves, but may also damage neurons by sequestering the protein, preventing it from functioning normally in the cell.23,24
Currently, much remains unknown about the neurodegenerative mechanisms involved in ALS. However, extensive research into the genetic products involved in ALS pathogenesis has identified numerous molecular and cellular processes, including oxidative stress, axonal/cytoskeletal dysfunction, endoplasmic reticulum stress, mitochondrial dysfunction, glia-mediated neuroinflammation, and alterations to proteostasis and RNA metabolism.5 Although the latter 2 mechanisms appear to be the most relevant, numerous pathways are common to multiple ALS genes, indicating that the origin of the disease is multifactorial.5
This review analyses the accumulated evidence that astrocytes not only react to degenerative damage to neurons, but also participate significantly in the process of neurodegeneration. To that end, we analyse the role of these cells in the main genetic alterations causing ALS. It should be noted that although the sporadic form of ALS is not hereditary, due to the nature of its etiology, some patients with this form of the disease may present certain glial cell behaviours, described below.
Astrocytes in SOD1-ALSThe SOD1 enzyme is present in various cell compartments, including the cytoplasm, nucleus, mitochondrial intermembrane space, peroxisomes, and lysosomes; this enzyme is expressed ubiquitously and abundantly in all tissues. Its function is detoxification, catalysing the conversion of the free radical superoxide into water or hydrogen peroxide25; loss of this function leads to the death of motor neurons secondary to altered mitochondrial function and axonal transport.25,26 The SOD1 gene is involved in practically all known putative pathways linked to the pathogenic mechanisms of ALS, including mitochondrial and/or axonal dysfunction, endoplasmic reticulum and/or oxidative stress, neuroinflammation, and proteostasis.15
The second most common hereditary cause of ALS is autosomal dominant mutations to SOD1, which account for 20% of familial cases.27 Rat and mouse models expressing mutant forms of murine or human SOD1, like patients with ALS, present progressive degeneration and selective death of motor neurons, gliosis, and ubiquitinated inclusions of misfolded proteins.28
Numerous studies of transgenic animal models and human patients report reduced expression of the EAAT2/GLT-1 glutamate transporter.29–31SOD1-mutant astrocytes can release high levels of D-serine, an endogenous coagonist of NMDA receptors; this favours the excitotoxicity hypothesis of ALS, due to the intensification of glutamate toxicity observed in motor neurons in in vivo models.32,33
Furthermore, mutant SOD1 loss in astrocytes delays disease onset and prolongs the early stage of disease progression.34 Similarly, it has been possible to extend the survival and slow the degeneration of motor neurons with transplantation of glial-restricted precursor cells (capable of differentiating into astrocytes) into the spinal cord of SOD1G93A rats (a transgenic rat model that generates mutant SOD1 protein with dismutase activity). On the other hand, wild-type rats presented increased motor neuron death and motor dysfunction after transplantation of astrocyte precursor cells with mutant SOD1.35 Together, these findings suggest that reactive astrocytes contribute to neurotoxicity in the pathogenesis of ALS in SOD1 rodent models.4
In astrocytes with mutant SOD1 and human phenotype, loss of function has been reported to induce similar effects, including the release of neurotrophic factors, reduced supply of lactate (involved in homeostasis), and reduced protection of neurons against glutamate-induced cytotoxicity. Curiously, motor neuron survival significantly improved when these functions were restored.15,30,36–38
In experimental models, astrocytes present a proinflammatory phenotype in ALS associated with SOD1 mutation, as a result of reciprocal interaction with other non-neuronal cells.4 In the near future, we may be able to consider new treatment approaches based on astrocyte replacement, as it has been reported that transplantation of astrocyte precursor cells in wild-type rodents delayed progression of the disease and extended the survival time of the study animals.15
Another promising finding is the significant reduction in disease progression and increased survival times observed in a mouse model of ALS in which mutant SOD1 was specifically deleted in astrocytes and microglial cells.26,34,39
Astrocytes in C9orf72-ALSAlthough the functions of C9orf72 are not fully understood, it is known to be expressed in both the CNS and in peripheral tissues (spleen, lymphocytes, and bone marrow). Patients with ALS present GGGGCC repeat expansions of hundreds or thousands of copies (the normal range is generally ≤ 30 copies). Interestingly, patients with repeat expansions in this gene, both in sporadic and in familial ALS, often present inappropriate social behaviour, loss of empathy, disinhibition, obsessive-compulsive behaviour, abnormal eating patterns, and apathy.40 The gene is known to be involved in putative pathways linked to pathogenic mechanisms of ALS, such as neuroinflammation, trophic factors, and aberrant RNA metabolism.15
C9orf72 mutations, observed in 25%–40% of familial cases of ALS, are the most frequently observed variants, except in Asian populations.41 A study using cocultures of neurons and astrocytes derived from patients with ALS associated with C9orf72 or SOD1 mutations reported that, independently of whether they were from patients with sporadic or familial ALS, astrocytes were equally toxic in both genetic conditions, potentially indicating a common mechanism.42
It has also been shown that reduced expression of the SRSF1 protein (serine and arginine rich splicing factor 1), a nuclear export adaptor for the transportation of C9orf72 transcripts, prevents C9orf72-induced neurodegeneration of motor neurons in cocultures of neurons and astrocytes derived from patients with C9orf72-ALS, implicating the gene in astrocyte-mediated neurotoxicity.43
The available evidence suggests that both familial and sporadic ALS involve an altered metabolic profile, which is important in the CNS in times of bioenergetic stress. This alteration involves deficient metabolism of fructose, adenosine, and glycogen in C9orf72 induced astrocytes, which have been associated with increased mouse motor neuron toxicity in a coculture of induced astrocytes derived from neuron progenitor cells.44,45 Specifically, an in vitro study found that astrocyte-mediated neurotoxicity due to dysfunction of adenosine metabolism secondary to adenosine deaminase inhibition could be reduced with inosine supplementation, which significantly increased glycolytic energy production and, consequently, motor neuron survival.46
Extracellular vesicles may also play an important role in the neurotoxic phenotype of mutant C9orf72 astrocytes. In normal physiological conditions, astrocytes regulate neuron function and the formation and maintenance of synapses; part of this communication is regulated by secreted extracellular vesicles. The main component of these vesicles are microRNAs (negative regulators of gene expression), which are highly important in the communication between motor neurons and astrocytes in ALS.46 Recent studies have reported dysregulated extracellular vesicle formation and microRNA expression in astrocytes derived from patients with C9orf72-ALS, dramatically affecting axon/neurite length and motor neuron survival in vitro. Specifically, downregulation of miR-494-3p results in increased expression of semaphorin-3A, involved in axon growth and maintenance. Restoring levels of this microRNA results in negative regulation of semaphorin-3A in motor neurons, considerably increasing their survival in vitro.46
Current evidence from mutant C9orf72 models suggests that astrocytes acquire neurotoxic properties through several mechanisms, including energy imbalance and alterations in microRNA metabolism; however, further research is needed to fully understand the molecular mechanisms involved in this type of ALS.4
Astrocytes in TARDBP-ALSThe TARDBP gene was described as a component of ubiquitinated protein inclusions in the lesions observed in sporadic ALS and frontotemporal dementia.47,48 The gene encodes TDP-43, which binds nuclear RNA to approximately 6000 RNA targets in the brain, and regulates mRNA transport, transcription, stability, and splicing.40 Dysfunction of TDP-43 causes cell toxicity due to aggregation (misfolding) of the protein or due to total or partial loss of function (either as a result of aggregation or depletion of the protein in the nucleus), causing neurodegeneration.49
While ALS is characterised by motor neuron death, astrocytes contribute to neurodegeneration. In a transgenic rat model with mutant human TDP-43 restricted to astrocytes, selective expression of the protein was sufficient to cause non–cell-autonomous death of motor neurons due to deficiency of neuroprotective genes and induction of neurotoxic genes in astrocytes.49
Similarly, a transgenic mouse model with partial loss of function of TDP-43 demonstrated elimination of this protein, both in peripheral and in CNS tissues, suggesting that motor neurons, in particular, present increased susceptibility to TDP-43 dysfunction. Furthermore, as these transgenic mice mainly displayed TDP-43 knockdown in the astrocytes of the spinal cord, dysfunction of the protein seems to be a key factor in motor neuron degeneration.50
It has also been reported that TDP-43M337V fibroblasts from patients with ALS and cultures of TDP-43Q331K mouse astrocytes present reduced levels of the antioxidant glutathione, resulting in an impaired oxidative stress response in these cells due to the aberrant translation of key antioxidant proteins.51
Furthermore, TDP-43M337V astrocytes derived from patients with ALS present subcellular mislocalisation, elevated TDP-43 levels, and reduced cell survival, compared to control astrocytes.52 In a coculture with TDP-43M337V astrocytes, motor neuron survival was not affected; this demonstrates that these mutant astrocytes do not have a toxic effect on motor neurons.52
Despite the findings described above, the molecules responsible for neurodegeneration in TARDBP-ALS have not been fully identified in astrocytes; however, dysfunction of glial cells is probably related to the mutant TDP-43 protein.15
An important point to consider is the fact that deletion of TARDBP is lethal to the embryo; postnatal deletion also leads to rapid death of the animal. Therefore, it seems unlikely that silencing of this gene can precisely model all aspects of TDP-43 alterations in ALS.53,54 However, primary astrocytes overexpressing TDP-43 or lacking the TARDBP gene showed no toxic effect on wild-type motor neurons in cocultures or after transplantation into the spinal cord of wild-type rats.55,56 It is unclear whether astrocytes without mutant TDP-43 aggregates are a reliable model of ALS.
Astrocyte-mediated excitotoxicity in ALSAstrocytes play an important role in numerous neurodegenerative diseases. One essential function of these cells is to maintain homeostasis of extracellular glutamate concentrations in the synaptic cleft, as excessively high levels cause neuron hyperactivation, increasing the deregulated influx of calcium; this results in neurotoxicity and, subsequently, neuronal death.57 Glutamate is a neurotoxic component involved in inflammation in ALS; however, the available evidence shows that D-serine induces glutamate excitotoxicity in motor neurons.32
In reactive astrocytes, intracellular Ca2+ concentration is increased, promoting the release of glutamate, which acts on motor neuron synapses and on AMPA and NMDA receptors. In ALS, AMPA receptors lack the GluR2 subunit, favouring influx of Ca2+ through the receptor channel. These astrocytes are unable to produce EAAT2 transporters, and the excitotoxicity phenomenon continues, with severe repercussions.5
In the 1990s, studies both in animal models and in humans yielded the first data on glial dysfunction in ALS, examining the role of astroglial glutamate transporters. These studies showed a considerable reduction in EAAT2/GLT-1 expression in astrocytes of the motor cortex and spinal cord in sporadic and familial ALS, both in human patients and in SOD1 rodent models.30,58 Similarly, the first in vitro and in vivo studies found that specific molecular elimination of this protein can cause degeneration and paralysis of neurons, including motor neurons.31 It was subsequently reported that the spinal cord in TDP-43M337V transgenic mice also exhibits a progressive reduction in GLT-1 and GLAST transporters.49
However, inflammatory mediators and proinflammatory cytokines play an important role in astrocyte-mediated toxicity. With this in mind, interferon-stimulated genes (GBP2, IFI27l2A, IFI44, IFIT1, IFIT3, ISG15, and USP18) in SOD1G93A transgenic mice presented positive regulation, specifically in astrocytes surrounding motor neurons in the spinal cord at presymptomatic ages. Furthermore, partial silencing of the interferon alpha receptor 1 inhibited the interferon signalling pathway and increased the life expectancy of SOD1G93A mice, compared to SOD1G93A mice with total silencing of the receptor.59 Together, these results show that positive regulation of interferon-stimulated genes in astrocytes may be related to the dysfunction of these cells in the spinal cord of mutant SOD1 mice; in particular, partial silencing of the interferon alpha receptor 1 gene is relevant to the survival of these animals.59
Transforming growth factor (TGF) is a cytokine involved in numerous biological functions, including immune homeostasis and the neurotrophic response. Changes in the TGF-β signalling pathway play a role in ALS, and have been shown to be correlated with the progression of reactive astrocytosis in cocultures of motor neurons and astrocytes from SOD1G93A mice.60 In the same model of ALS, neurons also showed impairment of the different signalling pathways affected in astrocytes (complement, coagulation cascade, cytokine–cytokine receptor interaction, focal adhesion, actin cytoskeleton regulation, extracellular matrix–receptor interaction), demonstrating that the presence of mutant SOD1 protein induces similar changes in both cell types, via similar signalling pathways. Therefore, astrocytes and motor neurons severely affect one another, demonstrating a profound, dynamic intercellular communication in the presence of SOD1G93A.60
Finally, it has been reported that in ALS, astrocytes present other dysfunction, such as a reduced capacity to release the metabolic substrate lactate, as well as mitochondrial defects.37,61 Degenerating astrocytes were first observed in presymptomatic stages, when living motor neurons showed axonal damage.62 With respect to this degeneration, glutamatergic alterations are harmful both to motor neurons and to astrocytes. Specifically, the metabotropic glutamate receptor 5 (mGluR5) has been shown to have a gliotoxic effect, whereas mGluR5 antagonists have a protective effect, reducing astrocytic degeneration, delaying ALS onset, and prolonging the survival of SOD1G93A mutant mice.63
Future perspectivesA growing body of evidence shows the pivotal role of glial cells in various physiological and pathological processes in ALS. Due to the different interconnected processes coordinated by these cells, it is unsurprising that acute and chronic CNS insults not only cause neuronal damage, but also trigger a range of complex responses that play a critical role in the progression of the disease. Therefore, glial cells represent an important therapeutic target in numerous disorders of the CNS.
In recent years, the application of gene editing technologies has redefined treatment strategies in numerous neurodegenerative diseases, aiming to correct genetic defects, modulate neuroinflammatory pathways in glial cells, and promote the conversion of astrocytes to neurons and oligodendrocytes. Therefore, the use of gene therapy to correct different mutations in glial cells is a fundamental tool in the treatment of different genetic and sporadic diseases of the CNS, including ALS.
ConclusionALS is a fatal, progressive neurodegenerative disease. Due to its heterogeneity and mainly idiopathic nature, it is particularly challenging to develop potential treatments to improve or increase patients’ quality of life.
Until a few decades ago, glial cells in the CNS were thought to function merely as a support. Today, new functions continue to be attributed to glial cells in maintaining neuron/glia homeostasis, as well as an important role in the pathophysiology of multiple neurodegenerative diseases, including ALS.
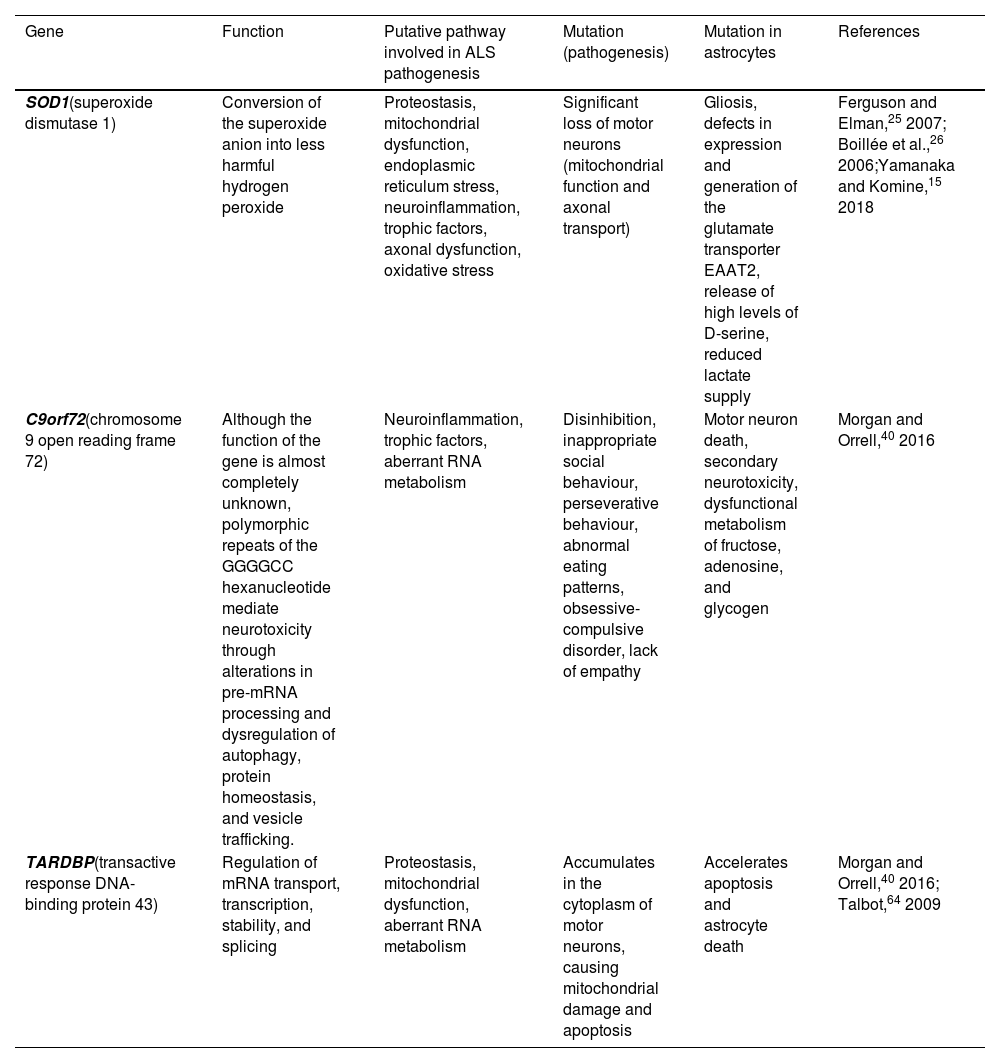
In particular, astrocytes play a key role in ALS pathogenesis and motor neuron death due to gain/loss of function and acquired toxicity. Mutations in the SOD1, C9orf72, and TARDBP genes are pathologically representative of the majority of known states within ALS; this study addresses how astrocytes present key functional and molecular differences as a result of the specific mutation (Table 1).
Genetic mutations associated with amyotrophic lateral sclerosis, and the alterations they cause in astrocytes.
| Gene | Function | Putative pathway involved in ALS pathogenesis | Mutation (pathogenesis) | Mutation in astrocytes | References |
|---|---|---|---|---|---|
| SOD1(superoxide dismutase 1) | Conversion of the superoxide anion into less harmful hydrogen peroxide | Proteostasis, mitochondrial dysfunction, endoplasmic reticulum stress, neuroinflammation, trophic factors, axonal dysfunction, oxidative stress | Significant loss of motor neurons (mitochondrial function and axonal transport) | Gliosis, defects in expression and generation of the glutamate transporter EAAT2, release of high levels of D-serine, reduced lactate supply | Ferguson and Elman,25 2007; Boillée et al.,26 2006;Yamanaka and Komine,15 2018 |
| C9orf72(chromosome 9 open reading frame 72) | Although the function of the gene is almost completely unknown, polymorphic repeats of the GGGGCC hexanucleotide mediate neurotoxicity through alterations in pre-mRNA processing and dysregulation of autophagy, protein homeostasis, and vesicle trafficking. | Neuroinflammation, trophic factors, aberrant RNA metabolism | Disinhibition, inappropriate social behaviour, perseverative behaviour, abnormal eating patterns, obsessive-compulsive disorder, lack of empathy | Motor neuron death, secondary neurotoxicity, dysfunctional metabolism of fructose, adenosine, and glycogen | Morgan and Orrell,40 2016 |
| TARDBP(transactive response DNA-binding protein 43) | Regulation of mRNA transport, transcription, stability, and splicing | Proteostasis, mitochondrial dysfunction, aberrant RNA metabolism | Accumulates in the cytoplasm of motor neurons, causing mitochondrial damage and apoptosis | Accelerates apoptosis and astrocyte death | Morgan and Orrell,40 2016; Talbot,64 2009 |
Although no cure for ALS has yet been developed, improving patient survival and quality of life fundamentally depends on multidisciplinary care, nutrition, and symptomatic treatment, together with pharmacological interventions. Therefore, future studies should address the function of astrocytes, taking into account different therapeutic approaches with potential to be highly beneficial for patients with ALS.
Numerous questions remain unanswered with respect to the aetiology of ALS; however, astrocytes have gradually become “rising stars,” offering great promise for the future treatment of this neurodegenerative disease.
FundingDB-A is supported by CONACYT-Mexico fellowship510113.
Informed consentThe authors declare that no patient data appear in this article.
Ethical considerationsThe authors declare that no human or animal experiments were conducted as part of this study.
