



En 1832, Thomas Hodgkin describió en 7 pacientes una enfermedad primitiva de los ganglios linfáticos diferente a los procesos reactivos ocasionados por alteraciones inflamatorias. De estos casos, dos correspondieron a sífilis y uno a linfosarcoma. Actualmente la enfermedad de Hodgkin se considera un proceso linfoproliferativo maligno, caracterizado por la presencia en el tumor de células de Reed-Sternberg. En el presente trabajo se aborda la sintomatología, diagnóstico, estadios y tratamiento de la enfermedad de Hodgkin.
La enfermedad de Hodgkin tiene con frecuencia un comienzo ganglionar y se extiende inicialmente a través del sistema linfático y con posterioridad por vía sanguínea. Este tipo de extensión y la disponibilidad de tratamientos eficaces determinan que sea curable el 75-80% de los pacientes.
En la actualidad se desconoce la causa y el mecanismo directo de esta enfermedad, aunque existen datos sobre la presencia del virus de Epstein-Barr sobre fenómenos de inmunodepresión con asociación de virus y VIH, y ciertas anomalías citogenéticas, así como la asociación de enfermedad de Hodgkin con otros virus.
La enfermedad de Hodgkin es poco frecuente. Representa el 1% de todas las neoplasias malignas e incide con una frecuencia de 3-3,5 nuevos casos por 100.000 habitantes y año. La curva de incidencia según la edad es típicamente bimodal, con un pico en los adultos jóvenes entre 15 y 35 años y otro después de los 55 años. El pico se desplaza a edades tanto más juveniles cuanto menor es el nivel socioeconómico del área estudiada. En conjunto hay un predominio evidente en los varones, con cifras proporcionales de hasta 1,5/1 aproximadamente. Con todo, en los adultos jóvenes la incidencia se iguala prácticamente en ambos sexos, al ser en esta edad más frecuente la variedad esclerosis nodular que predomina en el sexo femenino.
Anatomía patológica
El diagnóstico de enfermedad de Hodgkin sólo debería hacerse con rotundidad cuando se hallan células de Reed-Sternberg típicas. Incluso en la variedad esclerosis nodular, en la que el hallazgo de células lacunares dentro de un contexto típico puede ser ya suficiente criterio diagnóstico, continúa siendo idónea tal exigencia. En cambio, y una vez establecido el diagnóstico, cuando se trata de precisar la extensión de la enfermedad a otros órganos, ya no se requiere la presencia obligada de células de Reed-Sternberg y puede bastar el hallazgo de otros criterios cito o histológicos suficientemente expresivos para admitir la nueva localización.
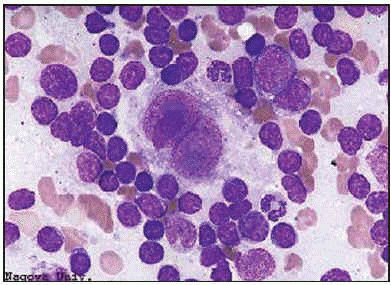
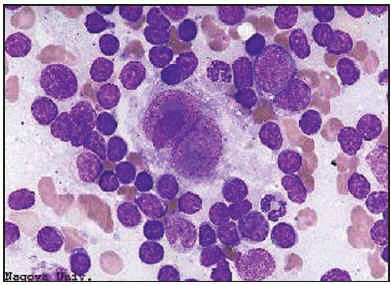
La célula de Reed-Sternberg típica es una célula gigante de 15 o 40 µm con citoplasma amplio, que suele ser directamente basófilo. El núcleo es, por lo general, grande, a veces múltiple y a menudo con lobulaciones. Lo más característico es la binucleación, con dos elementos nucleares de forma arriñonada que, confrontados, dan una imagen en espejo. En el centro de cada núcleo o lóbulo aparece un gran nucléolo, que suele ser único y ocupa más de la mitad del núcleo. En la tinción de May-Grünwald-Giemsa los nucléolos aparecen azulados, confiriendo a la célula un aspecto muy característico.
Esta célula de Reed-Sternberg paradigmática es la que se encuentra en la celularidad mixta (CM). En la práctica, con todo, sólo se halla como elemento gigante predominante en 1 de cada 5 pacientes, por lo que es mucho más frecuente que dominen las otras variantes de célula de Reed-Sternberg. Así, en la esclerosis nodular (EN), que es el tipo más a menudo observable en los países occidentales, pues se da en 3 de cada 4 pacientes, la variante es la célula «lacunar», con uno o varios núcleos que, dotados de pequeños y a veces nucléolos basófilos, menos destacados que en la variedad típica, se hallan rodeados por un citoplasma pálido y retraído por los fijadores habituales. Estas células lacunares coexisten con un número variable, a veces bajo, de células de Reed-Sternberg típicas.
En el predominio linfocítico (OL) las células diagnósticas son diferentes de la célula de Reed-Sternberg clásica. Se trata de células gigantes con núcleos «en palomita de maíz» (pop-corn cells) o L y H (linfocito e histiocito), variantes de células de Reed-Sternberg con núcleo arrugado, doblado y multilobular, dotado de pequeños nucléolos usualmente basófilos y con escaso citoplasma basófilo.
En la depleción linfocítica (DL), si bien se halla con frecuencia células de Reed-Sternberg típicas, suele dominar la proliferación de las llamadas células variantes mononucleares (células de Hodgkin), que siendo también grandes y dotadas de un nucléolo muy destacado pueden inducir a confusión con un linfoma no Hodgkin.
El diagnóstico de enfermedad de Hodgkin sólo debería hacerse con rotundidad cuando se hallan células de Reed-Sternberg típicas
Con la aplicación de los anticuerpos monoclonales se ha configurado un fenotipo inmunológico relativamente característico de las células de Reed-Sternberg. En efecto, la células de Reed-Sternberg típicas de la variedad CM y las equivalentes de las variedades EN y DL poseen un fenotipo propio de las células linfoides activadas con presencia de los antígenos CD30 (Ki-1), CD25 (IL-2), CDw70 (Ki-24), CD71 (receptor de transferrina) y HLA-DR. Es también muy positivo el marcador granulocítico CD15 (Leu M1), mientras que es típicamente negativo el CD45 (antígeno leucocitario común) que, por otra parte, es habitualmente positivo en los linfomas no hodgkianos. Con todo, y aunque raras veces, el CD45 puede ser positivo y el CD15 negativo. De todas maneras, la presencia de células tumorales CD15 positivas es altamente sugestiva de enfermedad de Hodgkin. Con todo, en un 20% de los casos el CD15 es negativo. Para evitar equívocos cabe añadir que en la célula de Reed-Sternberg pueden encontrarse marcadores B (CD19, CD20 o CD22), ocasionalmente marcadores T (CD2, CD3 o CD4) o ninguno de ellos. En la variedad PL y en concreto en su subtipo más frecuente, que es el nodular, las células de Reed-Sternberg llamadas «L» y «H» muestran un comportamiento atípico, al ser CD15 y CD30 negativas y CD20, CD45 y EMA (antígenos de la membrana epitelial, que suele ser negativo en las células de Reed-Sternberg típicas) positivos, en un fondo de predominio linfocítico B, distante del T que predomina en los otros tipos.
Síntomas y signos
Los síntomas y signos se relacionan principalmente con la localización, el número y la extensión de las masas ganglionares implicadas. La mayoría de los pacientes se presentan con adenopatías cervicales y mediastínicas, pero sin síntomas sistémicos. A medida que la enfermedad se disemina por el sistema mononuclear fagocítico, generalmente a localizaciones contiguas, se desarrollan otras manifestaciones. La velocidad de progresión varía según el subtipo histopatológico. Puede aparecer precozmente un prurito intenso; a menudo hay fiebre, sudación nocturna y pérdida de peso cuando están afectados ganglios internos (retroperitoneal o mediastínicos voluminosos), vísceras (hígado) o médula ósea. En ocasiones se observa fiebre de Pel-Ebstein (algunos días de fiebre elevada que alternan regularmente con días o semanas de temperatura normal o inferior a la normal). Un síntoma de mecanismo poco claro que puede aportar una clase diagnóstica precoz es el dolor inmediato en las regiones afectadas tras ingerir bebidas alcohólicas.
La afección ósea suele ser asintomática, pero puede producir dolor con lesiones osteblásticas vertebrales (vértebras de «marfil») y, raras veces, lesiones osteolíticas con fracturas por compresión. La pancitopenia se debe en ocasiones a la invasión de la médula ósea, en general en la variedad de depleción linfocítica. La invasión epidural que comprime la médula espinal puede ocasionar paraplejía. El síndrome de Horner y la parálisis laríngea pueden ser el resultado de la presión ejercida por los ganglios linfáticos aumentados de tamaño sobre los nervios simpáticos cervicales y recurrente laríngeo, respectivamente. Los dolores neurálgicos son consecuencia de la compresión de las raíces nerviosas. Raras veces aparecen lesiones intracraneales, gástricas y cutáneas y, en caso de estar presentes, sugieren enfermedad de Hodgkin asociada al VIH.
La obstrucción de los conductos biliares intrahepáticos o extrahepáticos por masas tumorales produce ictericia. El edema en las piernas puede ser consecuencia de la obstrucción linfática en la pelvis o la ingle. La compresión traqueobronquial puede causar disnea intensa y sibilancias. La infiltración del parénquima pulmonar puede simular una consolidación lobular o una bronconeumonía y originar cavitación o abscesos pulmonares.
La mayoría de los pacientes presenta un trastorno lentamente progresivo de la inmunidad retardada o celular (función de la célula T), que contribuye en la enfermedad avanzada a la aparición de infecciones bacterianas frecuentes y, más raramente, de infecciones por hongos, virus y protozoos. La inmunidad humoral (producción de anticuerpos) o función de las células B también está deprimida en la enfermedad avanzada. La caquexia es habitual y los pacientes fallecen frecuentemente por sepsis.
Diagnóstico
El diagnóstico de la enfermedad de Hodgkin es anatomopatológico y debe realizarse con las técnicas habituales y con el inmunofenotipo para clasificar las distintas formas histopatológicas. El diagnóstico diferencial se establece prioritariamente con los LNH. El linfoma B mediastínico tiene células que se asemejan a las de Reed-Sternberg, los linfomas anaplásicos pueden confundirse con la enfermedad de Hodgkin tipo depleción linfocitaria, y la enfermedad de Hodgkin tipo predominio linfocitario se considera en la actualidad como un linfoma de fenotipo B.
Otras enfermedades que pueden plantear problemas de diagnóstico diferencial con la enfermedad de Hodgkin son las adenopatías de tumor primario desconocido, la mononucleosis, la sarcoidosis, la toxoplasmosis y el síndrome de Sjögren.
Estadios de la enfermedad
La radioterapia, la quimioterapia o combinaciones de ambas son curativas, pero para alcanzar este objetivo debe determinarse la extensión del «estadio patológico». Para que el tratamiento tenga éxito se requiere de forma crítica determinar la extensión de la enfermedad, que puede estar inicialmente localizada (estadio I o II) en el 50% de los pacientes. El sistema de estadiaje de Ann Arbor es el que se emplea con mayor frecuencia.
Estadio I. Limitado a una región ganglionar linfática anatómica.
Estadio II. Dos o más regiones ganglionares linfáticas en el mismo lado del diafragma.
Estadio III. Enfermedad en ambos lados del diafragma que afecta a ganglios linfáticos o al bazo.
Estadio IV. Afección extranodal (médula ósea, pulmón o hígado).
Subclasificación
Afectación extranodal adyacente a un ganglio linfático afectado: por ejemplo, la enfermedad que cursa con adenopatías cervicales y adenopatía hiliar con infiltración pulmonar adyacente se clasifica en estadio IIE y no como estadio IV.
Los estadios se clasifican como clínicos (CS) o patológico (PS) y son aún más definidos por la ausencia (A) o la presencia (B) de síntomas constitucionales (pérdida de peso, fiebre o sudación nocturna). La presencia de B suele asociarse a una gran masa de la enfermedad.
La modificación de Cotswold de los estadios de Ann Arbor utiliza X para designar una localización voluminosa (mayor a un tercio del diámetro torácico o mayor a 10 cm de diámetro).
Los procedimientos no invasivos de clasificación incluyen la TC de tórax, abdomen y pelvis y la gammagrafía con galio. La gammagrafía ósea y la RM no suelen ser necesarias. La linfografía bipedal puede estar indicada en pacientes con TC abdominal y pélvica normales. Los estudios clínicos que intentan detectar la enfermedad infradiafragmática proporcionan falsos positivos o negativos en el 25-33% de los pacientes, por lo que debe valorarse la realización de una laparotomía que incluya esplenectomía, biopsia de los ganglios linfáticos mesentéricos o retroperitoneales (especialmente los aumentados de tamaño en la TC o la linfografía) y biopsias de la médula ósea y del hígado, cuando influya de manera significativa en las decisiones terapéuticas. Sin embargo, las indicaciones de la laparotomía de clasificación se han reducido notablemente en los últimos años, de manera que sólo se plantea en pacientes en estadio clínico IIA o menor y en quienes se considera la irradiación en mantle. Si el paciente va a recibir quimioterapia, no se precisa la laparotomía de clasificación.
Tratamiento
Los avances terapéuticos en la enfermedad de Hodgkin permiten en el momento actual una esperanza de curación a 20 años del 75% de los pacientes, con una toxicidad cada vez menor en cuanto a la quimioterapia y a la radioterapia y una reducida morbilidad al ser muy limitadas las indicaciones de la laparotomía. Las dos armas terapéuticas más importantes son la radioterapia y la quimioterapia.
Radioterapia
Representó la primera posibilidad de curación de la enfermedad de Hodgkin en sus formas localizadas. La aplicación de la radioterapia se realiza en zonas afectadas y adyacentes. La erradicación del tumor depende de la dosis y se necesitan 35-40 Gy en los lugares invadidos y 30-35 Gy en las zonas clínicamente no afectadas, administrados a una dosis de 1,5-2 Gy/día durante 5 días a la semana.
Quimioterapia
La combinación de agentes alquilantes y de alcaloides de la Vinca proporcionan un número importante de respuestas completas (65%). El MOPP (mecloretamina, vincristina, procarbazina, prednisona), introducido en 1967, permitió un 80% de respuestas completas con la mitad de curaciones. Sobre el protocolo MOPP se han introducido diversas variables con la finalidad de intentar mejorar su eficacia o reducir su toxicidad. En la actualidad no se utiliza al haberse suprimido la mecloretamina.
Los avances terapéuticos en la enfermedad de Hodgkin permiten en el momento actual una esperanza de curación a 20 años del 75% de los pacientes, con una toxicidad cada vez menor en cuanto a la quimioterapia y a la radioterapia
El protocolo ABVD (adriamicina, bleomicina, vinblastina, dacarbazina) tiene como base el sinergismo de sus fármacos, la eficacia de las antraciclinas en linfomas y la utilización de fármacos no contenidos en el programa MOPP. Sobre esta base se combinaron ambos tratamientos de forma alternativa, lo que permitió reducir la toxicidad de la mecloretamina o fusionarlos formando un protocolo híbrido, denominado MOPP/ABV. El protocolo MOPP/ABV se basa en la hipótesis de Goldie-Coldman, según la cual la exposición del tumor a siete fármacos diferentes en 8 días reduce la producción de resistencia celular originada por el crecimiento del tumor. Este protocolo no es superior al ABVD y es más tóxico.
Los protocolos NOVP (mitoxantrona, vincristina, vinblastina, prednisona) y VBM (vinclastina, bleomicina, metotrexato) son tratamientos que se han utilizado en estadios iniciales asociados a la radioterapia, y han demostrado ser eficaces y poco tóxicos, sobre todo el NOVP.
Indicaciones terapéuticas y resultados
Estadios I y II
Actualmente se evita la práctica de la laparotomía para el estudio de la extensión en la enfermedad de Hodgkin. En los enfermos con adenopatías cervicales altas, sin masa tumoral grande, sin lesión mediastínica y con histología de predominio linfocitario, rara vez la extensión es mayor que la detectada de forma clínica. Sólo en los estadios IA estudiados con laparotomía las posibilidades de curación exclusivamente con radioterapia son próximas al 90% de los casos, utilizando un campo de radiación tipo manto. En los estadios II tratados con radioterapia en manto las posibilidades de recaída son del 20-35%, la supervivencia a los 10 años es del 78% y la supervivencia libre de enfermedad es del 65-75% según las series estudiadas.
La presentación de la enfermedad de Hodgkin con tumor mediastínico aparece en más de la mitad de los casos. En un número importante de éstos la masa es mayor de 10 cm de diámetro o superior a un tercio del diámetro máximo del tórax. En estos casos, las recaídas con tratamiento exclusivo mediante radioterapia llegan al 50% y la masa mediastínica abultada es el factor pronóstico más significativo de los estadios I y II.
En los estadios IB y IIB tratados con radioterapia y con estadio determinado mediante laparotomía, las recaídas son del 48% a los 7 años y la supervivencia alcanza el 57%.
Como conclusión, la posibilidad de curación de la enfermedad de Hodgkin en estadios I y II a los 10 años es del 80% y la supervivencia del 90% de los casos.
Actualmente es excepcional el estudio mediante laparotomía del estadio en la enfermedad de Hodgkin. Por lo común se realizan tres ciclos de quimioterapia con los protocolos referidos y radioterapia tipo manto o incluso campo afectado. La quimioterapia menos tóxica y con similar eficacia es el protocolo NOVP, que no produce alopecia, náuseas ni vómitos, no es leucemógeno y sólo origina mialgias de poca intensidad. Estas combinaciones consiguen más del 90% de respuestas completas y una supervivencia libre de enfermedad del 85% en estos pacientes.
Estadios III y IV
El tratamiento fundamental de estos estadios es la quimioterapia, sobre todo en los estadios III2A, IIIB y IV. El protocolo considerado durante mucho tiempo como el más eficaz ha sido el MOPP. Existe la comprobación de que, debido a la mostaza nitrogenada, produce una importante e irreversible toxicidad, que origina azoospermia con esterilidad permanente en la totalidad de los pacientes y leucemogénesis.
Los niños deben tratarse con quimioterapia tipo ABVD, que produce menos esterilidad que el MOPP, y con radioterapia con menos dosis y campos más reducidos
Los protocolos tipo ABVD consiguen mejores resultados y tienen menos toxicidad que la administración exclusiva de MOPP. Las posibilidades de curación son aproximadamente del 50-65% de los casos. Las respuestas completas con ABVD son del 83% y la supervivencia libre de enfermedad a los 5 años es del 61%. Los resultados parecen algo superiores con MOPP/ABV híbrido. Se administran 6 ciclos de uno u otro de los protocolos, separados por un período de 28 días. En los casos en que queda masa tumoral residual o había una masa previa de tamaño importante se tratan esas lesiones con radioterapia.
Resistencias al tratamiento y recaídas
Aproximadamente el 15% de los pacientes muestran resistencia primaria, que se expresa por la no obtención de respuesta completa con quimioterapia. Las posibilidades de curación en estos casos con quimioterapia de rescate son mínimas y por lo tanto debe recurrirse a intensificación con altas dosis de quimioterapia y trasplante de progenitores hematopoyéticos (TPH). Las recaídas de los pacientes con enfermedad de Hodgkin después de haber obtenido una respuesta completa es del 30%, de los cuales el 40-50% recaen en el primer año. Las recaídas durante el primer año tienen mal pronóstico y su índice de curación con tratamientos de rescate es inferior al 10%.
Intensificación y trasplante de progenitores hematopoyéticos
Su indicación es clara en las siguientes situaciones:
Resistencias iniciales al tratamiento. Se puede lograr una curación del 25-30%.
Recaídas durante el primer año después de haber obtenido respuesta completa. Los resultados, si sólo existe ese factor pronóstico (recaída en el primer año), son del 50-60% de curaciones.
Recaídas después del primer año de haber obtenido respuesta completa. El retraso en esta indicación aumenta la mortalidad y reduce la eficacia.
Tratamiento inicial en situaciones de muy mal pronóstico: estadios IV, con afección pulmonar y ósea.
Tratamiento de los niños
La enfermedad de Hodgkin en niños requiere el mismo tratamiento que en los adultos. Sin embargo, origina trastornos del crecimiento por irradiación de los huesos y alteraciones tiroideas que causan tumores de esta glándula. Los niños deben tratarse con quimioterapia tipo ABVD, que produce menos esterilidad que el MOPP, y con radioterapia con menos dosis y campos más reducidos y hay que evitar la laparotomía con esplenectomía, ya que puede facilitar sepsis graves. *