




Objetivos: Los objetivos fueron identificar y clasificar las causas y los patrones de crecimiento de la microcefalia adquirida (o progresiva) y buscar hipotéticas correlaciones entre las causas, los patrones de crecimiento y el cociente de desarrollo/CI.
Métodos: Mediante una revisión retrospectiva de la historia clínica y la curva de crecimiento, con evaluaciones formales del cociente de desarrollo o del CI (n = 34), estudiamos a 51 niños (24 chicos) de 0,7 a 11,3 años de edad con un perímetro occipitofrontal temprano por encima del segundo percentil y los posteriores por debajo (DE –2,03).
Resultados: Las causas pudieron clasificarse en 5 grupos: idiopáticas, familiares, sindrómicas, sintomáticas y mixtas. Identificamos cuatro patrones de crecimiento cefálico: tipo A, disminución inicial desde valores normales a menos del segundo percentil, seguido de un crecimiento inferior y paralelo al segundo percentil; tipo B, disminución sostenida, alejándose del segundo percentil; tipo C, disminución por debajo del valor normal, con posterior recuperación parcial; tipo D, datos insuficientes. En 12 niños hubo disminuciones concomitantes en los percentiles de peso y en 5 de ellos también en los de talla. Los lactantes con menores puntuaciones z del perímetro craneal al final del estudio también mostraron menores puntuaciones z del peso final y de la talla final. No hubo correlación entre grupo causal y patrón de crecimiento. Los valores del cociente de desarrollo/CI fueron principal-mente < 100 y no mostraron correlación con la puntuación z del perímetro craneal, la causa o el patrón.
Conclusiones: La clasificación de los grupos causales y del patrón de crecimiento debería ayudar al manejo clínico. Ni la causa ni el patrón predijeron el resultado. Las asociaciones con una escasa ganancia ponderal y un mal crecimiento corporal merecen más estudios.
La microcefalia adquirida (también conocida como progresiva) es una alteración en la que el perímetro craneal de un niño está en los valores normales al nacer y durante un período indefinido posterior, pero luego no aumenta con la rapidez normal y, en consecuencia, cruza los percentiles hasta bajar del segundo percentil. Según nuestra experiencia, es una forma relativamente habitual de microcefalia. Aun cuando existe una amplia bibliografía sobre la microcefalia, es escasa sobre este subtipo. Las causas reconocidas incluyen la lesión cerebral adquirida y el síndrome de Rett, el síndrome de Angelman, el síndrome de Down y otros síndromes menos frecuentes, pero provocan la minoría de los casos1,2. También existen distintos patrones de crecimiento cefálico de los niños con microcefalia, que no han sido estudiados. En consecuencia, en la actualidad carecemos de información que ofrecer a la mayoría de las familias cuyos hijos presentan microcefalia adquirida.
Para ayudar a remediar esta situación, realizamos un estudio de observación en el que intentamos clasificar los patrones y, cuando fue posible, las causas de la microcefalia adquirida. Pese a ser un estudio explorador, también supusimos la existencia de una relación entre la causa y el patrón, de forma que las causas lesionales más graves se asociarían con un fracaso de crecimiento más intenso, y que el fracaso de crecimiento más intenso se asociaría con menores puntuaciones del cociente del desarrollo (CD)/CI.
MÉTODOSFue un estudio retrospectivo de niños derivados a una consulta de neurología pediátrica durante un período de 10 años, entre una población local ¿500.000. Los profesionales de la asistencia sanitaria tomaron las mediciones en los marcos clínicos habituales, incluyendo las exploraciones de alta neonatal por médicos en formación, las mediciones ambulatorias y las mediciones comunitarias por enfermeras domiciliarias o de otro tipo. En todos los casos con causa sintomática (v. la definición más adelante), el patrón de crecimiento craneal fue la causa de la derivación, con o sin preocupación por el cierre temprano de la fontanela anterior.
La microcefalia adquirida se definió por un perímetro craneal temprano al nacer por encima del segundo percentil con mediciones posteriores por debajo de este límite. El segundo percentil corresponde a –2,03 DE por debajo de la media o una puntuación z de –2,03. Las gráficas de percentiles y los datos de crecimiento fueron los más modernos para los niños británicos3. Están basados en datos transversales de referencia de Reino Unido y se aplican desde el nacimiento hasta los 16 a 18 años de edad.
La causa derivó de las evaluaciones diagnósticas clínicas habituales, incluyendo la medición del perímetro craneal de la familia inmediata, y las investigaciones específicas de cada niño. Los niños con retraso del desarrollo o retraso mental inexplicables se sometieron, al menos, a la determinación del cariotipo, del estado de X frágil, el estudio de la función tiroidea y las mediciones de los valores de aminoácidos, glucosaminoglucanos y creatina cinasa (en los chicos). El patrón de crecimiento se identificó a partir de la inspección visual de los diagramas de percentiles.
Se invitó a los niños a realizar un estudio neuropsicológico con las Bayley Scales of Infant Development4, las Wechsler Preschool and Primary Scales of Intelligence-Revised5 o la Wechsler Intelligence Scale for Children6, según fuera adecuado a su grupo de edad. Todas las puntuaciones se presentan como CI o CD, para facilitar las comparaciones entre las distintas escalas (puntuación media 100; DE 15). Un CD/CI < 70 se calificó como retraso del desarrollo/retraso mental.
Muchos de los demás datos se presentan como estadística descriptiva. Se utilizaron tests t apareados para comparar las puntuaciones z iniciales del perímetro craneal con las del final del estudio. Se calcularon las diferencias medias con intervalos de confianza del 95%. Se controlaron las diferencias respecto a la normalidad mediante la elaboración de un histograma. Todas las diferencias apareadas se compararon de esta manera. Las diferencias entre las causas de microcefalia adquirida se compararon mediante el análisis de la variancia, tras controlar los residuos respecto a la normalidad. Cuando se detectaron diferencias significativas se aplicó el test post hoc de Scheffé. Se aceptó un valor arbitrario de significación estadística del 5% (bilateral). Según la práctica estadística habitual, informamos de los valores exactos de p. El análisis de los datos se realizó con SPSS 10.0 (SPSS, Chicago, IL). El estudio fue aprobado por el comité de Ética local de South Sheffield.
RESULTADOSEncontramos 51 niños (24 chicos y 27 chicas) (mediana de edad 4,5 años [límites 0,7-11,3 años]). Dos habían nacido pretérmino (33 y 35 semanas), y el resto a término. Las mediciones inicial y final se tomaron en las medianas a los 0,03 y 4,49 años de edad, respectivamente. Cuarenta y nueve niños mostraron de 1 a 6 mediciones por encima del segundo percentil a edades comprendidas entre 2 semanas y 3 años, antes de desarrollar microcefalia. Dos niños con mediciones normales sólo al nacer tuvieron unas puntuaciones z iniciales de –0,55 y –1,25.
Las causas se dividieron inicialmente en subgrupos múltiples, pero la revisión final permitió simplificarlos en 5 grupos: 1) idiopático, es decir, no se pudo reconocer una causa a partir de la historia, la exploración física o los exámenes complementarios, y no hubo otros hallazgos anormales; 2) familiar, es decir, uno de los padres, un hermano, o ambos tuvo un perímetro craneal en o por debajo del segundo percentil; 3) sindrómico, es decir, con anomalías asociadas; 4) sintomático, es decir, posterior a un acontecimiento patogénico, o 5) mixto, es decir, ≥2 de los antes mencionados. En el grupo idiopático, 6 niños estaban en condiciones por lo demás nor-males, por lo que no se sometieron a otra investigación, aparte de una RM cerebral en un caso, que mostró una disminución del volumen de la sustancia blanca con gliosis. Dos niños, con CI de 63 y 71, se sometieron a las investigaciones antes enumeradas con resultados nor-males; el primero también se sometió a una RM cerebral con resultados normales.
En el grupo familiar (n = 12), los familiares afectados fueron los siguientes: madre y hermano (incluyendo 2 parejas de hermanos), n = 4; sólo la madre, n = 4; sólo un hermano, n = 2; sólo una hermana, n = 1; la madre y una hermana, n = 1. Los fenotipos de los demás miembros de la familia afectados no se registraron con mayor detalle porque no estaban incluidos en el estudio. En la mayoría de los casos, la microcefalia sólo fue reconocida al tomar las mediciones a la familia tras la identificación del caso índice.
En el grupo sindrómico (n = 13), 4 pacientes mostraron síndromes reconocidos (síndrome de Rett, n = 2; síndrome de Gómez, López y Hernández, n = 1; microdeleción del cromosoma 9q34.3, n = 1). Los dos últimos niños fueron asignados a este grupo antes de identificar el síndrome. Los otros 9 niños fueron asignados a este grupo porque mostraron rasgos dismórficos, anomalías congénitas, o ambas características. Uno tuvo también anemia drepanocítica. Por la selección de la derivación no se incluyó en el estudio a ningún niño con síndrome de Down.
En el grupo sintomático (n = 15), las causas individuales fueron las siguientes: prenatales: infección por el virus de Epstein y Barr con alteraciones de la señal de la sustancia blanca en la RM; traumáticas; abuso materno de drogas; gliosis de la sustancia blanca; perinatales: encefalopatía neonatal7 (n = 7), leucomalacia periventricular/parto pretérmino; posnatales: hemorragia intracraneal, deshidratación hipernatrémica, fracaso global del crecimiento. Este último paciente fue el único en el que los parámetros de peso y talla disminuyeron antes que el perímetro craneal.
Todos los casos del grupo mixto mostraron un componente familiar (2 madres y 1 hermana). Dos pacientes presentaban rasgos sindrómicos y uno padecía una diplejía asociada con una vaginitis bacteriana durante el embarazo, varios episodios de amenaza de parto pretérmino y alteraciones de la señal de la sustancia blanca en las imágenes T2 de la RM.
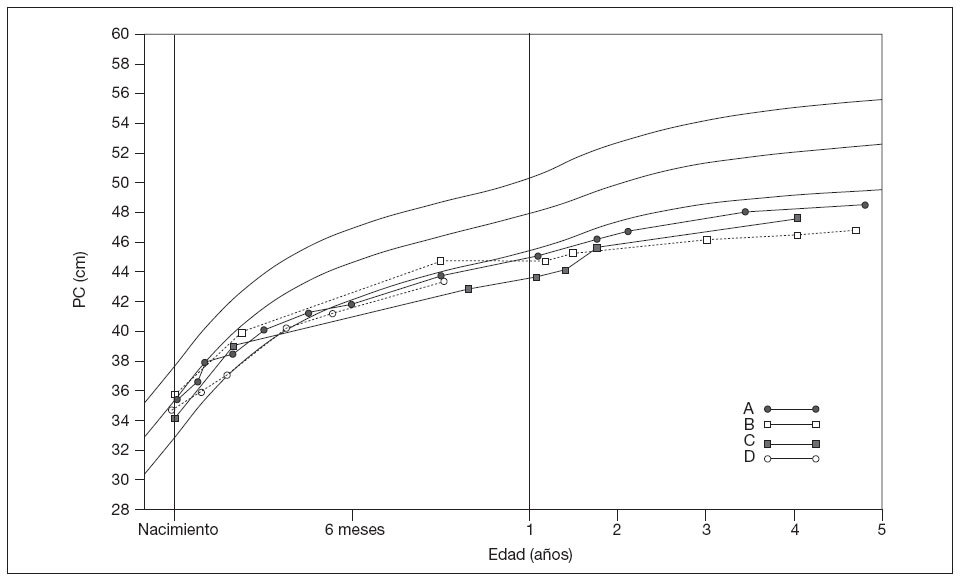
Las observaciones visuales de los patrones llevaron a una clasificación en 4 grupos (fig. 1). En el tipo A (n = 30), la disminución inicial desde valores normales a me-nos del segundo percentil se siguió por un crecimiento constante, inferior y paralelo al segundo percentil. En el grupo B (n = 12) se produjo una disminución constante que se alejaba del segundo percentil. En el tipo C (n = 4), tras una disminución por debajo de los valores normales se produjo una recuperación parcial, con microcefalia persistente. En el tipo D (n = 5) no hubo datos suficientes, habitualmente por la corta edad. Siete niños mostraron una disminución coetánea del percentil de peso, pero no del de talla; otros 5 niños presentaron disminuciones de los percentiles de peso y de talla (tabla 1).
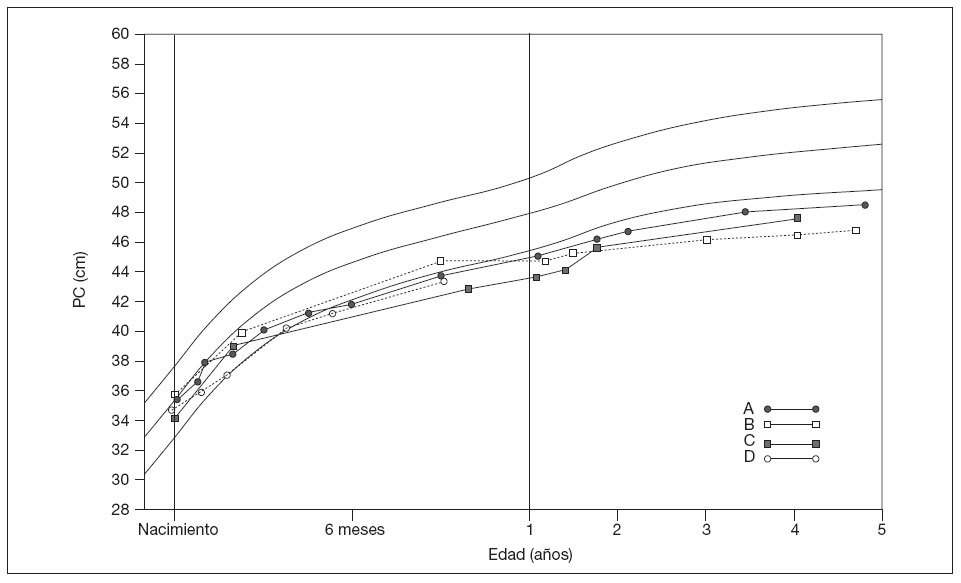
Fig. 1. Ejemplos compuestos que muestran los patrones de crecimiento en las niñas. Las líneas finas continuas indican el 2.º, 50.º y 98.º percentil. Los datos de crecimiento se utilizaron con autorización de la Child Growth Foundation3. PC: perímetro craneal.
TABLA 1. Análisis descriptivo según el grupo causal, que muestra las mediciones inicial y final de perímetro craneal, los patrones de crecimiento, los hallazgos de RM y los resultados de CD/CI

La tabla 1 presenta más detalles según la causa. La media de la puntuación z inicial del perímetro craneal del grupo familiar pareció ser menor que la de los demás grupos, aunque esta diferencia no fue significativa (p = 0,11). Las puntuaciones z del perímetro craneal final de todos los grupos fueron mucho más próximas, sin diferencias evidentes (p = 0,83). No hubo diferencias claras entre los grupos respecto a la edad en que el perímetro craneal cayó por debajo del segundo percentil. No se realizó la RM craneal a todo el grupo, especialmente en los niños pequeños que hubieran necesitado anestesia general, porque esta investigación no se consideró éticamente aceptable si el niño no mostraba hallazgos clínicos anormales. Con esta limitación, hubo cierta correlación entre los resultados de la RM y los grupos causales, ya que los niños con causas sintomáticas solieron presentar resultados anormales en la RM, mientras que los niños con causas familiares tenían una RM normal. Las alteraciones de la sustancia blanca consistieron en una disminución de volumen, gliosis periventricular, o ambos hallazgos. También hubo hallazgos lesionales o malformativos.
La tabla 2 ofrece más detalles según los patrones del crecimiento cefálico. Se observaron todos los patrones en todas las causas, pero los pacientes con el patrón B presentaron una causa sintomática más a menudo que los pacientes con el patrón A (6 de 12 pacientes frente a 8 de 30 pacientes). Los pacientes con el patrón B tuvieron unas puntuaciones z al final del estudio significativamente menores (p ≤ 0,0001) que los demás pacientes.
TABLA 2. Análisis descriptivo según el patrón de crecimiento, mostrando las mediciones inicial y final del perímetro craneal, los grupos causales, los hallazgos de la RM y los resultados de CD/CI

Los niños con mayores puntuaciones z en la situación inicial mostraron mayor puntuación z al final del estudio (diferencia media 2,96 [intervalo de confianza del 95% 2,66-3,26]; p < 0,0001). Las puntuaciones z del perímetro craneal no difirieron entre los chicos y las chicas al inicio del estudio (p = 0,28), pero sí al final de él (chi-cos, media –3,24; chicas, media –3,81; p = 0,36). No hubo relación entre la puntuación z final del perímetro craneal y la edad a la que se midió (p = 0,61). Doce de los 51 niños también mostraron patrones anormales de crecimiento del peso y la talla. La disminución del percentil de peso habitualmente coincidió con la del perímetro craneal. Los niños con menores puntuaciones z del perímetro craneal al final del estudio también mostraron menores puntuaciones z del peso final (p = 0,05) y la talla final (p = 0,038).
Diez de los 15 niños del grupo sintomático presentaron parálisis cerebral (nivel I de la Gross Motor Function Classification Scale, n = 3; nivel II, n = 1; nivel III, n = 1; nivel IV, n = 1; nivel V, n = 5), además de un niño del grupo familiar (nivel III) y otro del grupo sindrómico (nivel IV). Treinta y cuatro familias accedieron a realizar la evaluación de los CD o CI. En estos niños, la mediana de CD/CI fue 63. Sólo 3 (9%) de los 34 niños tuvieron una puntuación ≥100. El grupo idiopático tuvo la máxima mediana de puntuación (83) y el grupo sindrómico la mínima (45) (tabla 1). Los niños con patrón A tuvieron una mediana de puntuación superior a los niños con patrón B (tabla 2). La cifra de niños en estos grupos fue demasiado pequeña para poder realizar comparaciones estadísticas sensatas. La presencia o ausencia de anomalías asociadas del crecimiento no modificó estas puntuaciones. No hubo correlación entre CD/CI y la puntuación z del perímetro craneal medida simultáneamente (p = 0,4).
ANÁLISISÉste es el primer intento de estudiar sistemáticamente la microcefalia adquirida o progresiva2. Una razón importante fue ofrecer mejor información a los padres cuyos hijos desarrollaron la alteración. También esperábamos que fuese una ayuda diagnóstica útil para los médicos, porque la microcefalia adquirida fue el motivo de consulta de to-dos los niños, excepto los del grupo sintomático, en la que mayoritariamente siguió a su acontecimiento patogénico. Este último grupo también pudo distinguirse clínicamente por la presencia de otros signos neurológicos, como la parálisis cerebral. Este grupo corresponde a la microcefalia “adquirida” o “secundaria”; ambas son expresiones ambiguas, ya que se utilizan para distinguirla de la microcefalia primaria o genética y para distinguir el fracaso de crecimiento cerebral temprano del tardío. Nuestro estudio sugiere que muchos casos de aparición tardía probable-mente tengan una base genética. Como en los estudios de la microcefalia en conjunto, se reconoce una serie de causas1,2. Los intentos de clasificarlas no han desembocado en un método aceptado internacionalmente. Nuestra clasificación de este grupo tiene como ventajas ser práctica y sencilla.
Respecto a la causa, el grupo idiopático tuvo relativamente pocos casos. No habíamos esperado tantos casos familiares. Esto podría sugerir una “microcefalia familiar”, bastante similar a la macrocefalia familiar, pero la fuerte relación con la madre apunta a una influencia genética distinta. Nuestro poder de demostrar diferencias significativas fue muy limitado, y no podemos confirmar si este grupo también tuvo menor perímetro craneal inicial, comparado con los demás grupos. Los datos de RM disponibles indican que este grupo carece de anomalías estructurales. En el grupo sindrómico hubo muchos fenotipos distintos, la mayoría no reconocidos, y algunos con posible origen genético. En la microcefalia congénita hubo similares dificultades para clasificar casos probablemente sindrómicos7. Sin embargo, hubo muy pocos casos en el grupo de solapamiento/mixto. No hubo casos de síndrome de Down porque estos niños acuden a un servicio clínico específico y suelen presentar otros problemas. Es interesante que ningún niño tuviera sinostosis de las suturas, que raras veces se presenta con microcefalia adquirida. El síndrome de Gómez es la asociación de alopecia parietal, rombencefalosinapsis cerebelosa y sinostosis de las suturas; aunque nuestra paciente mostró una microcefalia adquirida, no presentaba sinostosis8. Esto sugiere que, en este síndrome, el patrón de crecimiento cefálico puede atribuirse incorrectamente a la sinostosis. Finalmente, la parálisis cerebral pareció tener mayor prevalencia en el grupo sintomático, lo que probablemente refleje las causas subyacentes.
También es el primer intento de clasificar los patrones de crecimiento en la microcefalia adquirida. Nuestra clasificación es sencilla y práctica. La mayoría de los casos corresponde al patrón A. Sin embargo, nos sorprendió la ausencia de una correlación evidente entre la causa y el patrón. Creíamos que el patrón B, con un fracaso de crecimiento más intenso, sólo se asociaría con causas sintomáticas pero, aunque el patrón B apareció con mayor frecuencia en este grupo, también se produjo en otros grupos. Como se esperaba, las puntuaciones z finales de los niños con patrón B fueron menores que las de los niños con patrón A, lo que refleja el más grave patrón de crecimiento de los primeros. El patrón C tampoco estuvo claramente relacionado con la causa. El crecimiento cefálico posnatal se atribuye tradicionalmente al desarrollo de la sustancia blanca y la mielinización en la primera infancia y al crecimiento del hueso craneal en la segunda infancia y la adolescencia. En los niños pequeños, el perímetro craneal se correlaciona estrechamente con el volumen cerebral. Esto significa que la microcefalia adquirida es atribuible principalmente a un fracaso de crecimiento de la sustancia blanca. Los datos de la RM, de un limitado número de nuestros pacientes, apoyan hasta cierto punto esta sugerencia, ya que muchos pacientes muestran anomalías del volumen o la señal de la sustancia blanca. Sin embargo, en muchos pacientes no fue así, y no realizamos análisis volumétricos sistemáticos. Se ha sugerido que la microcefalia prenatal refleja un menor número de neuronas, mientras que la microcefalia adquirida refleja un menor número de conexiones dendríticas y sinápticas9. El patrón A (que, por ejemplo, es típico en las chicas con síndrome de Rett) indica también que distintos procesos pueden controlar los períodos temprano y tardío del crecimiento cerebral posnatal.
Tampoco esperábamos que tantos pacientes mostrasen una disminución del percentil de peso iniciado al mismo tiempo que la caída del percentil cefálico, la mayoría de ellos sin un descenso simultáneo del percentil de talla. Esto no pudo atribuirse a una privación energética, en la que el fracaso del aumento ponderal suele preceder a la disminución de la talla, afectándose por último el crecimiento cefálico. Esta asociación no ha sido reconocida anteriormente, pero no pudimos encontrar una explicación. Este grupo merece más estudios.
Los valores de CD/CI de los niños con microcefalia adquirida estuvieron por lo general por debajo de la media, cerca de la mitad estaba en la parte inferior de los valores normales (70-100) y el resto < 70. No encontramos relación con el perímetro craneal. Esto concuerda con los resultados de la microcefalia congénita, en la que existe un CI muy inferior a la media, pero sin una clara correlación con el tamaño10. De forma similar, en los supervivientes de la UCIN no hubo una estrecha correlación entre el resultado del desarrollo y el tamaño cefálico absoluto o corregido11. No pudimos detectar factor de predicción alguno, aunque nuestra capacidad estuvo limitada por lo pequeño de las cifras. Aunque las mediciones del perímetro craneal se tomaron de forma no normalizada, estudios anteriores mostraron la buena reproducibilidad de estas mediciones entre los observadores12.
Las proporciones de cada grupo causal pueden variar en una población de estudio, ya que este estudio estuvo limitado por ser retrospectivo y selectivo. Contamos con datos de otros 7 niños pero, como no pudimos comunicarnos con los pacientes, nuestro comité de Ética negó el permiso de utilizar estos datos. Esta exclusión no modifica las proporciones de cada grupo ni nuestras conclusiones.
Nuestro estudio trató de examinar el patrón en el que el tamaño cefálico está en los límites normales al nacer y posteriormente cae por debajo de estos límites. Sin embargo, la mediana del tamaño cefálico ya estuvo por debajo de la media al inicio, lo que indica un probable origen prenatal en la mayoría de los casos, y los valores finales se correlacionaron con los valores iniciales. Así pues, la microcefalia adquirida es una entidad un tanto artificial que se basa únicamente en una presentación clínica específica. La definición también excluyó a los niños cuyo perímetro craneal declinó a través de los percentiles pero acabó por encima del segundo percentil, lo que pudo ocurrir en algunos niños, por ejemplo, cerca de la mitad de los niños con síndrome de Down13. Optamos por utilizar el segundo percentil por estar más próximo a –2 DE y por disponer de los datos en consultas con gráficas de crecimiento estándar, mientras que otros argumentaron que –2,5 o –3 DE es más adecuado, especialmente en los estudios genéticos7. En los niños con patrón B, la edad a la medición afectará este punto.
El pequeño tamaño de la muestra (n = 51 en la situación inicial) implicó la limitación de los análisis estadísticos. No pareció sensato realizar un modelo estadístico complejo, y nos centramos en la comparación de las medias de grupo. El poder estadístico para comparar los grupos fue limitado, especialmente en las comparaciones de las causas de microcefalia adquirida. Por desgracia, será difícil estudiar más detenidamente en Reino Unido las preguntas planteadas en este grupo, porque durante la infancia ya no se mide rutinariamente el perímetro craneal.
CONCLUSIONESEste primer estudio de la microcefalia adquirida ha desembocado en una clasificación sencilla de los grupos causales y los patrones de crecimiento, que debería ayudar al manejo clínico. Es probable que muchos casos tengan origen genético, y parece existir una influencia materna en el grupo familiar. Ni el grupo causal ni el patrón de crecimiento predijeron el CD/CI. Las asociaciones con la mala ganancia ponderal y crecimiento corporal merecen más estudios.
AGRADECIMIENTOSEste trabajo fue financiado por el Sheffield Children’s Hospital Charity.
Damos las gracias a las familias participantes, a la Dra. Barbara Steele por su ayuda en la recogida y el análisis de los datos, al Dr. Jerry Wales por la transformación de los datos de crecimiento en puntuaciones z y a Vanda Cupit por su apoyo de secretaría.
Correspondencia: Peter Stuart Baxter, MD, Ryegate Centre, Sheffield Children’s NHS Foundation Trust, Tapton Crescent Road, Sheffield S10 5DD, Inglaterra.
Correo electrónico: peter.baxter@sch.nhs.uk