





El concepto de autoinflamación aparece por primera vez en la literatura médica en el año 1999, siendo acuñado por el Dr. Daniel L. Kastner del National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS)1. Su grupo identificó en aquella época los defectos genéticos responsables de la fiebre mediterránea familiar (FMF) y la fiebre hiberniana familiar (FHF; posteriormente renombrada como síndrome periódico asociado al receptor i del TNF-TRAPS), 2 enfermedades hereditarias con semejanzas clínicas y caracterizadas por episodios recurrentes de fiebre, serositis estéril y manifestaciones inflamatorias sistémicas2,3. Estos episodios inflamatorios agudos pueden ser desencadenados por factores inespecíficos tales como estrés, pequeños traumatismos o infecciones leves. Sin embargo, ya a finales de los años 90, se había demostrado que estos episodios agudos no eran debidos ni a la presencia de infecciones ni a procesos neoplásicos ni estaban asociados con la presencia de autoanticuerpos circulantes a títulos elevados, excluyéndose de una manera razonable las causas infecciosas, tumorales y autoinmunes para los mismos3,4.
Desde aquel año 1999 hasta nuestros días han tenido lugar importantes avances en múltiples áreas del conocimiento de estas enfermedades que básicamente se podrían resumir en los siguientes puntos:
El número de enfermedades autoinflamatorias hereditarias cuya base genética ha sido identificada ha aumentado hasta un total de 15 diferentes en la actualidad4–15. Además, muy probablemente este número continuará aumentando durante los próximos años como consecuencia de la aplicación de modernas herramientas de genética molecular.
Gracias a la identificación del gen defectuoso en cada una de las enfermedades, y consecuentemente de la proteína responsable, así como a los avances habidos durante los últimos 10 años en el conocimiento del sistema inmune innato, en la actualidad se conoce de una manera bastante detallada las bases fisiopatológicas subyacentes a cada una de estas enfermedades.
Se están aplicando en la práctica clínica diaria los estudios genéticos para alcanzar el diagnóstico definitivo de estas enfermedades hereditarias, de tal manera que se está vislumbrando que la diversidad clínica de cada una de ellas es mayor de lo descrito con anterioridad.
Se están administrando con éxito fármacos antiinflamatorios biológicos en pacientes afectos de diferentes enfermedades autoinflamatorias y esta administración se fundamenta en el conocimiento de la base fisiopatologica propuesta para cada una de ellas.
La estrecha relación existente entre las enfermedades autoinflamatorias y las células y moléculas del sistema inmune innato, puesta de manifiesto durante los pasados 10 años, han hecho necesaria una actualización del concepto de enfermedad autoinflamatoria. Así, en la actualidad se considera que las enfermedades autoinflamatorias son trastornos clínicos caracterizados por una inflamación anormalmente incrementada llevada a cabo fundamentalmente por las células y moléculas del sistema inmune innato con una predisposición por parte del individuo que la padece, entendiendo por tal predisposición tanto factores genéticos como ciertos efectos consecuencia de la interacción de factores ambientales con genes16 (fig. 1). Además, la definición actual de enfermedad autoinflamatoria es lo suficientemente amplia como para incluir tanto enfermedades hereditarias, con un clásico patrón mendeliano de herencia, como enfermedades complejas, sin una base genética clara o conocida (artritis idiopática juvenil de inicio sistémico, enfermedad de Still del adulto, síndrome PFAPA o síndrome de Marshall, artropatías microcristalinas [gota y seudogota], enfermedad de Behçet). Finalmente, también se ha propuesto la existencia de un espectro clínico continuo entre las enfermedades autoinflamatorias y las enfermedades autoinmunes17.
Clasificación de las enfermedades autoinflamatoriasClasificación clínica versus clasificación fisiopatológica
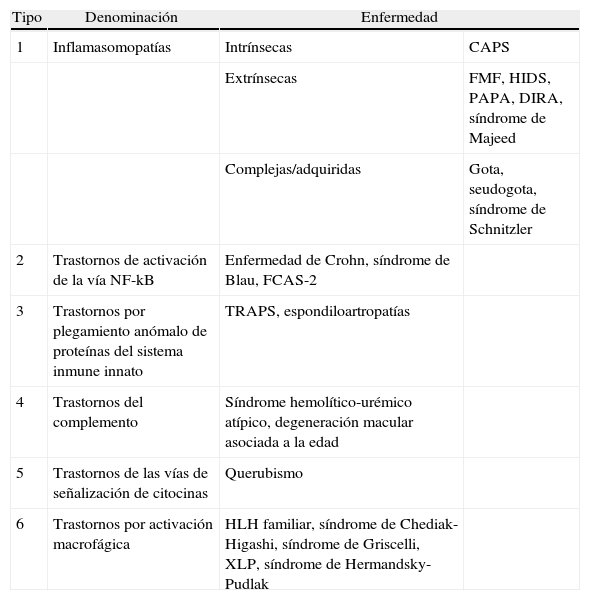
Debido a que el concepto de enfermedad autoinflamatoria fue propuesto en un contexto clínico la gran mayoría de clasificaciones aparecidas desde entonces han agrupado exclusivamente a las enfermedades autoinflamatorias monogénicas en las cuales un defecto genético heredable con un determinado patrón de herencia es la causa de la enfermedad. Sin embargo, el concepto de autoinflamación como mecanismo fisiopatológico puede estar presente, en mayor o menor medida, en un conjunto múltiple de enfermedades no necesariamente hereditarias con un patrón mendeliano clásico. Este hecho motivó una nueva propuesta de clasificación basada fundamentalmente en el mecanismo fisiopatológico principal propuesto para cada entidad que incluye grupos tales como enfermedades por disregulación de la vía de la interleucina 1 (IL-1), enfermedades por disregulación de la vía NF-kB o de las vías de transducción de señales de las citocinas, enfermdedades debidas a un plegamiento anómalo de las proteínas y enfermedades debidas a una activación macrofágica anormalmente incrementada4. En la tabla 1 se presenta la propuesta de clasificación fisiopatológica de estas enfermedades.
Propuesta de clasificación fisiopatológica de las enfermedades autoinflamatorias
| Tipo | Denominación | Enfermedad | |
| 1 | Inflamasomopatías | Intrínsecas | CAPS |
| Extrínsecas | FMF, HIDS, PAPA, DIRA, síndrome de Majeed | ||
| Complejas/adquiridas | Gota, seudogota, síndrome de Schnitzler | ||
| 2 | Trastornos de activación de la vía NF-kB | Enfermedad de Crohn, síndrome de Blau, FCAS-2 | |
| 3 | Trastornos por plegamiento anómalo de proteínas del sistema inmune innato | TRAPS, espondiloartropatías | |
| 4 | Trastornos del complemento | Síndrome hemolítico-urémico atípico, degeneración macular asociada a la edad | |
| 5 | Trastornos de las vías de señalización de citocinas | Querubismo | |
| 6 | Trastornos por activación macrofágica | HLH familiar, síndrome de Chediak-Higashi, síndrome de Griscelli, XLP, síndrome de Hermandsky-Pudlak | |
CAPS: síndromes periódicos asociados a la criopirina; DIRA: deficiencia del antagonista del receptor de IL-1; FCAS-2: síndrome autoinflamatorio familiar inducido por el frio tipo 2 o fiebre de Guadalupe; FMF: fiebre mediterránea familiar; HIDS: síndrome de hiper-IgD y fiebre periódica; HLH: linfohistiocitosis hemofagocítica; PAPA: síndrome de artritis piogénica estéril, pioderma gangrenoso y acné; TRAPS: síndrome periódico asociado al receptor i del TNF; XLP: síndrome linfoproliferativo ligado al cromosoma X.
Modificada de: Masters SL et al.4.
Desde finales de los años 90 se ha ido identificando la base genética de unas 15 enfermedades autoinflamatorias diferentes, todas ellas con un patrón de herencia mendeliano clásico. En conjunto, constituyen el grupo denominado enfermedades autoinflamatorias hereditarias. Sin embargo, el mecanismo fisiopatológico de la autoinflamación está presente en un conjunto de enfermedades mucho mayor en las cuales no se ha identificado una base genética subyacente (por no tenerla o por no haberse todavía identificado). Precisamente, es en este grupo de enfermedades autoinflamatorias no hereditarias donde en la actualidad se están ensayando los agentes bloqueantes de la IL-1, fármacos que ya han demostrado su eficacia clínica en el grupo de las hereditarias. En la tabla 2 se presenta una propuesta de clasificación de las enfermedades autoinflamatorias en función de la la base genética.
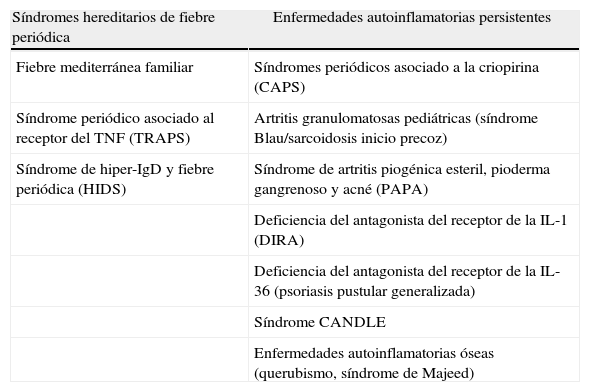
Propuesta de clasificación clínica de las enfermedades autoinflamatorias hereditarias
| Síndromes hereditarios de fiebre periódica | Enfermedades autoinflamatorias persistentes |
| Fiebre mediterránea familiar | Síndromes periódicos asociado a la criopirina (CAPS) |
| Síndrome periódico asociado al receptor del TNF (TRAPS) | Artritis granulomatosas pediátricas (síndrome Blau/sarcoidosis inicio precoz) |
| Síndrome de hiper-IgD y fiebre periódica (HIDS) | Síndrome de artritis piogénica esteril, pioderma gangrenoso y acné (PAPA) |
| Deficiencia del antagonista del receptor de la IL-1 (DIRA) | |
| Deficiencia del antagonista del receptor de la IL-36 (psoriasis pustular generalizada) | |
| Síndrome CANDLE | |
| Enfermedades autoinflamatorias óseas (querubismo, síndrome de Majeed) |
Dado que las principales enfermedades dentro del grupo de enfermedades autoinflamatorias hereditarias cursan en forma de episodios agudos, separados por intervalos asintomáticos, tiende a pensarse que todas estas enfermedades presentan este tipo de dinámica. Sin embargo, conforme se ha ido incrementando el número de enfermedades autoinflamatorias hereditarias se ha podido comprobar que algunas de ellas cursan de esta forma episódica, recibiendo el nombre global de síndromes hereditarios de fiebre periódica o recurrente, mientras que otras cursan de una manera fundamentalmente persistente. En la tabla 2 se indican qué enfermedades estarían incluidas en cada uno de estos grupos.
Inflamasoma e interleucina 1: papel central en la autoinflamaciónFamilia interleucina 1La IL-1 es la citocina de alarma del organismo prototípica ya que coordina la respuesta más temprana del sistema inmune en respuesta al daño bien sea de origen exógeno o bien de origen endógeno. La IL-1α y la IL-1β fueron los primeros miembros descritos de la familia IL-1 y los mejor conocidos. Sin embargo, esta familia IL-1 contiene en la actualidad un total de 11 miembros diferentes, estando la gran mayoría de ellos codificados por genes que se localizan en un cluster del brazo largo del cromosoma 218. En la tabla 3 se detallan los diferentes miembros conocidos de esta familia, así como los diferentes nombres que han recibido.
Miembros de la familia de la interleucina-1
| Nombre actual | Sinónimos | Mecanismo de acción |
| IL-1F1 | IL-1α | Agonista |
| IL-1F2 | IL-1β | Agonista |
| IL-1F3 | Antagonista del receptor de IL-1 (IL-1Ra) | Receptor antagonista |
| IL-1F4 | IL-18; factor inductor de IFN-γ | Agonista |
| IL-1F5 | Antagonista del receptor de IL-36 (IL-36Ra); FIL-1δ | Receptor antagonista |
| IL-1F6 | IL-36α; FIL-1¿ | Agonista |
| IL-1F7 | IL-1H4; IL-1ζ | Antiinflamatorio |
| IL-1F8 | IL-36β; IL-1H2 | Agonista |
| IL-1F9 | IL-36γ; IL-1¿ | Agonista |
| IL-1F10 | IL-1Hy2 | Receptor antagonista |
| IL-1F11 | IL-33 | Agonista |
Desde un punto de vista funcional, la IL-1α se sintetiza en su forma biológicamente activa pero la IL-1β lo hace en forma de una proproteína biológicamente inactiva (pro-IL-1β; isoforma 31 kDa). Para que esta proproteína se convierta en su forma activa (IL-1β; isoforma 17 kDa) debe ser degradada proteolíticamente por la caspasa-118,19. Debido a sus múltiples funciones biológicas la vía de la IL-1 presenta múltiples niveles de regulación, siendo algunos de los más importantes los señalados a continuación:
Procesamiento proteolítico de la pro-IL-1β, desempeñado fundamentalmente por una estructura conocida como inflamasoma de la que se hablará en el apartado siguiente.
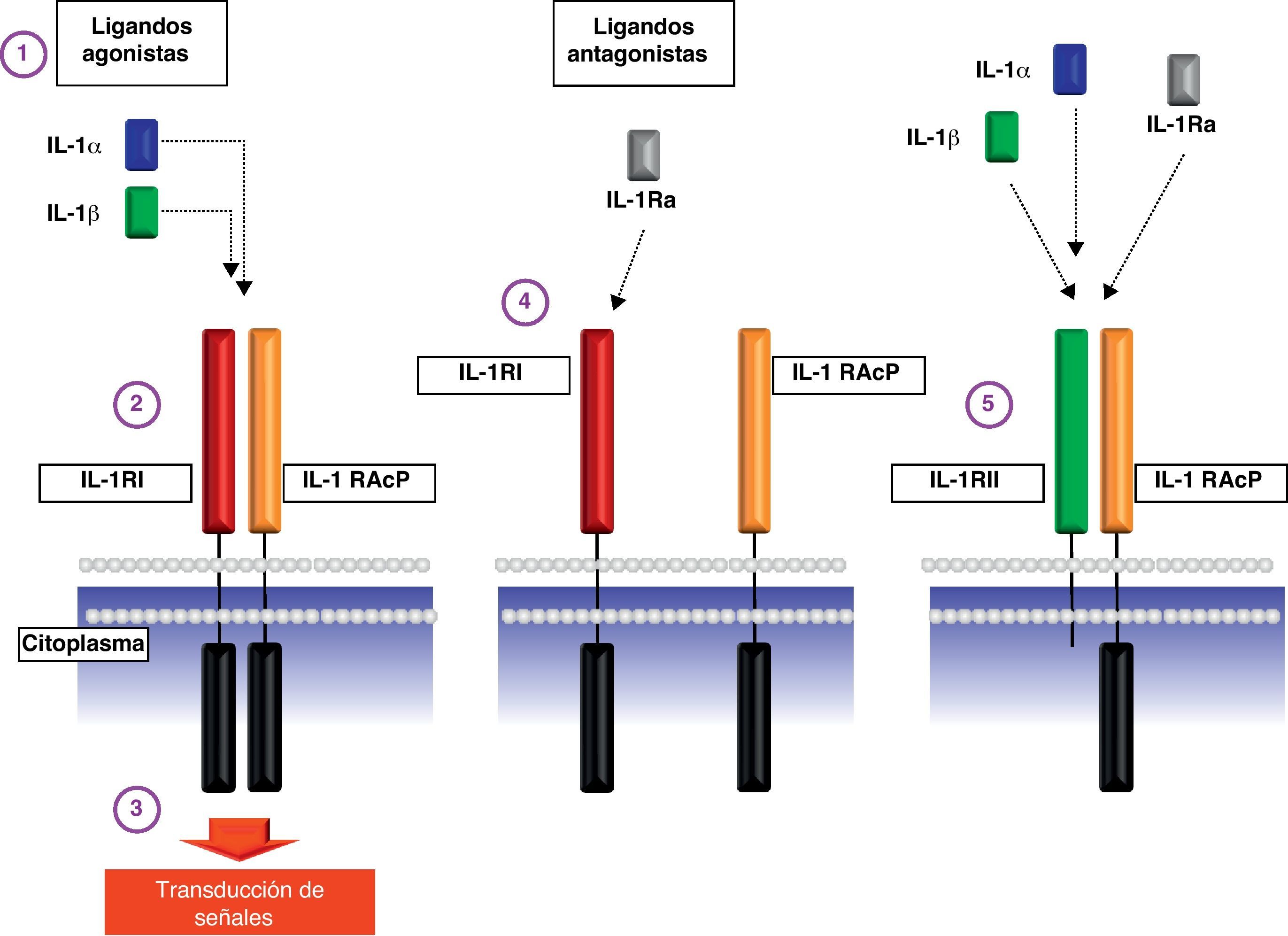
Las 2 citocinas activas (IL-1α y la isoforma 17 kDa IL-1β) se unen al receptor de la IL-1 de tipo i (transductor de señal al interior de la célula) y de tipo ii (no transductor de la señal por carecer de dominios intracelulares) (fig. 2).

Esquema de los receptores de membrana de la interleucina-1. Punto 1: Esquema de los ligandos agonistas IL-1α e IL-1β. Punto 2: Unión de los ligandos agonistas a la proteína IL-1RI y su posterior asociación con la proteína IL-1RAcP. Punto 3: Transducción de la señal proinflamatoria al interior de la célula mediante el acoplamiento de la proteína adaptadora MyD88 al dominio TIR del receptor de la IL-1. Punto 4: Unión del ligando antagonista IL-1Ra al receptor IL-1RI que impide tanto la asociación con la proteína IL-1RAcP como la transducción de señales intracelulares. Punto 5: Esquema del receptor decoy IL-1RII que puede unir tanto ligandos agonistas como antagonistas pero que no puede transmitir señales al interior de la celula por carecer de dominios citoplásmáticos. Abreviaturas: IL-1α: interleucina-1α; IL-1β: interleucina-1β; IL-1RI: receptor de tipo i de la interleucina-1; IL-1RAcP: proteína accesoria del receptor de IL-1; IL-1Ra: antagonista del receptor de IL-1; IL-1RII: receptor de tipo ii de la interleucina-1.
Cuando la citocina activa se une al receptor de IL-1 de tipo i se acopla la proteína accesoria del receptor de la IL-1 (IL-1RAcP) de tal manera que el complejo puede reclutar la proteína MyD88 e iniciar la transducción de señales al interior celular. Un miembro de la familia de IL-1, denominado antagonista del receptor de IL-1 (IL-1Ra), se une al receptor de tipo i de la IL-1 pero impide el reclutamiento de la proteína accesoria IL-1RAcP de tal modo que inhibe competitivamente la acción de las citocinas activas y de carácter agonista IL-1α y IL-1β (fig. 2).
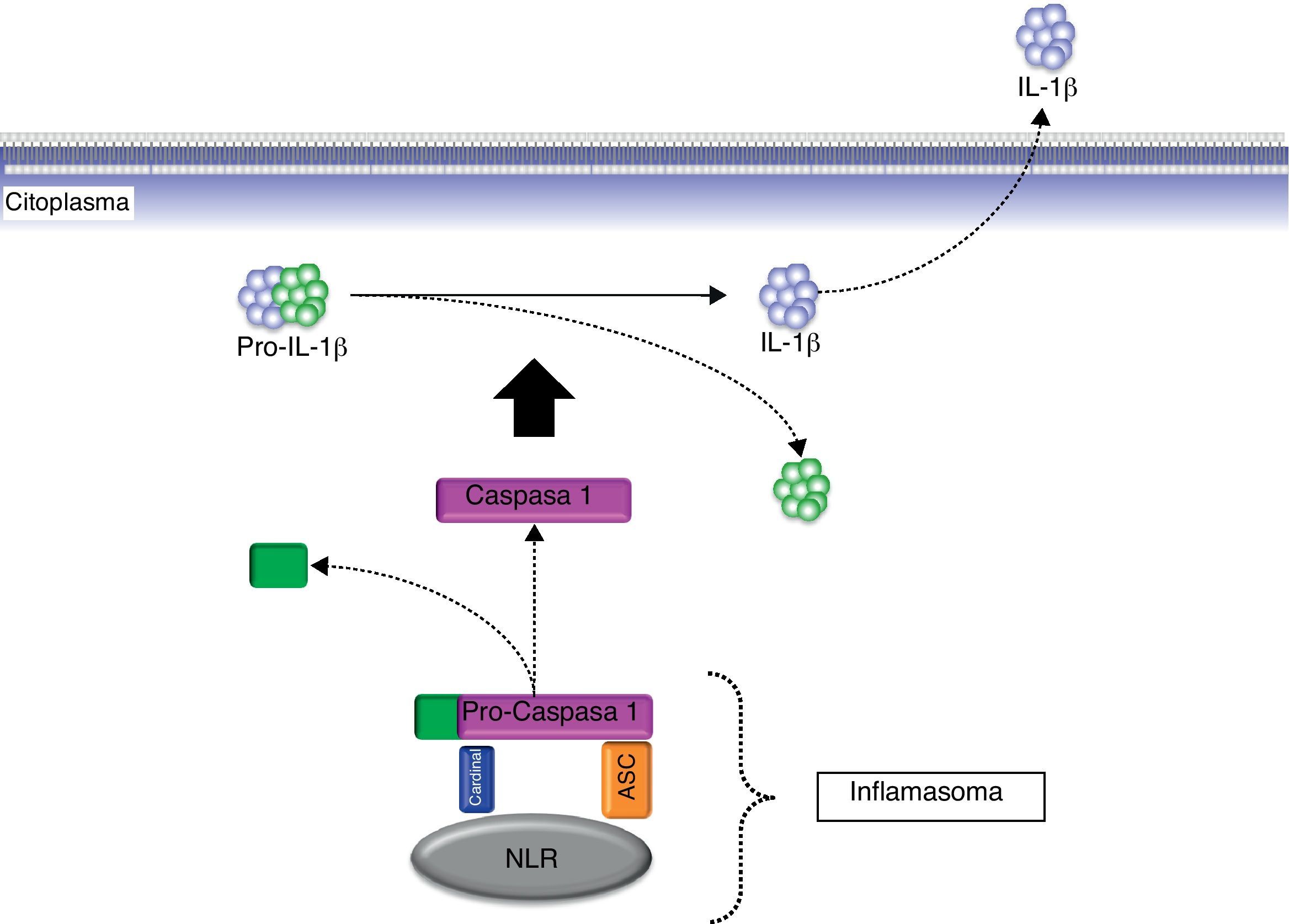
InflamasomaEn el año 2002, cuando se investigaba la función de la proteína criopirina mutada en los síndromes periódicos asociados a la criopirina (CAPS), se propuso el concepto de inflamasoma para señalar su relación con las caspasas proinflamatorias a semejanza del concepto ya existente de apoptosoma acuñado algún tiempo antes para hablar de las caspasas proapoptóticas20. El inflamasoma es un complejo citosólico no delimitado por membranas y compuesto por múltiples proteínas21. Asimismo, el inflamosoma es una estructura dinámica en cuanto que sus componentes se acoplan y desacoplan en función de la presencia o ausencia de determinados estímulos. Entre sus componentes estructurales figuran las caspasas proinflamatorias (caspasa-1, caspasa-5), ciertas proteínas acopladoras (ASC, Cardinal) y miembros de la familia de receptores NLR a la cual pertenece la criopirina21 (fig. 3). En cuanto que existen diferentes miembros de esta familia NLR, en sentido estricto, es lógico pensar que existen diferentes tipos de inflamasoma que pueden presentar una expresión tisular variable. En concreto, es el inflamasoma de criopirina (también denominado Nalp3-inflamasoma) el que desempeña un papel clave en la generación de la forma activa de las citocinas proinflamatorias IL-1β, IL-18 e IL-333.

Esquema del procesamiento de la pro-IL-1β biológicamente inactiva a la forma activa IL-1β mediante la acción del complejo multiproteico citosólico denominado inflamasoma. ASC: Apoptosis-associated speck-like protein containing a CARD; IL-1β: interleucina-1β; NLR; receptor de tipo Nod; pro-IL-1β: pro-interleucina-1β.
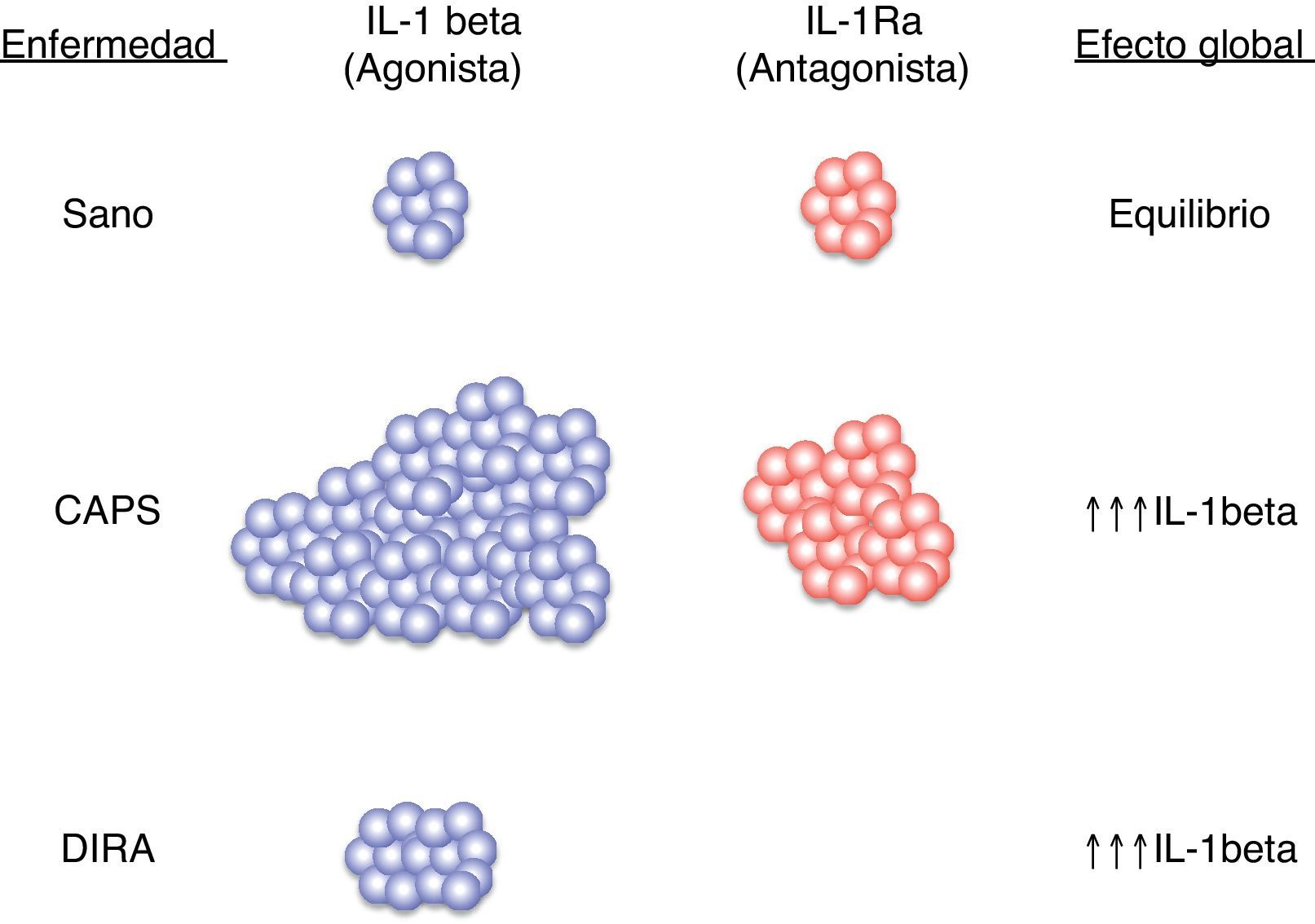
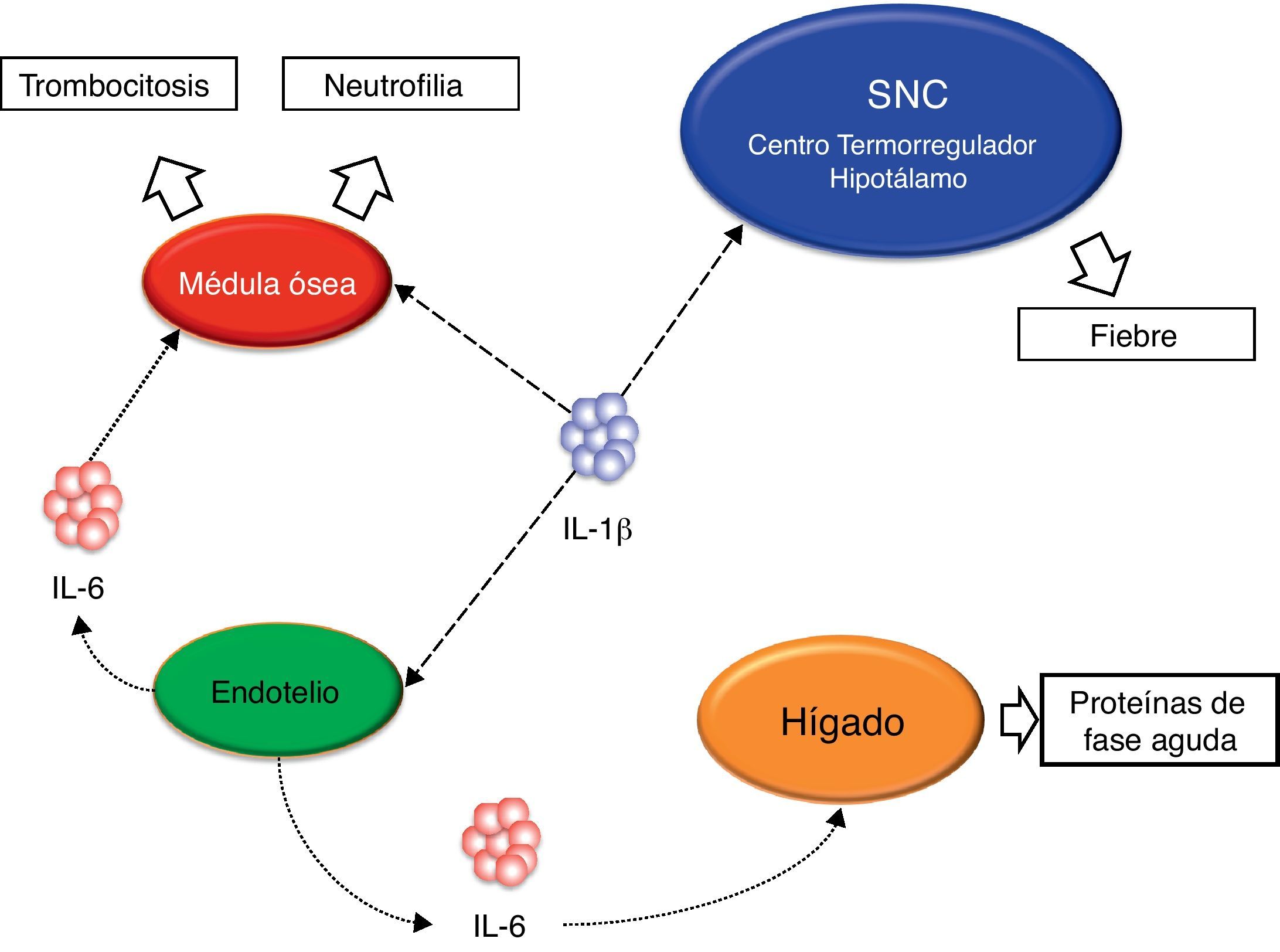
Diferentes estudios realizados con pacientes afectos de diferentes enfermedades autoinflamatorias han revelado en todos ellos una hiperfunción del Nalp3-inflamasoma que se traduce en una producción excesiva, y en ocasiones no regulada, de la citocina inflamatoria IL-1β22,23 (fig. 4). Precisamente, esta hiperproducción de IL-1β es considerada la responsable, directa o indirecta, de muchas de las manifestaciones observadas en estos pacientes tales como la fiebre, las manifestaciones cutáneas, el aumento de neutrófilos y plaquetas circulantes y el marcado aumento de las proteínas de fase aguda (proteína C reactiva, proteína sérica del amiloide, ferritina, factores del complemento, etc.)24,25 (fig. 5).
 en situación de normalidad y en diferentes enfermedades autoinflamatorias hereditarias. CAPS: síndromes periódicos asociados a la criopirina; DIRA: deficiencia del antagonista del receptor de IL-1; IL-1β: interleucina-1β; IL-1Ra: antagonista del receptor de IL-1.")
Esquema del equilibrio entre ligando agonista IL-1β y antagonista (IL-1Ra) en situación de normalidad y en diferentes enfermedades autoinflamatorias hereditarias. CAPS: síndromes periódicos asociados a la criopirina; DIRA: deficiencia del antagonista del receptor de IL-1; IL-1β: interleucina-1β; IL-1Ra: antagonista del receptor de IL-1.

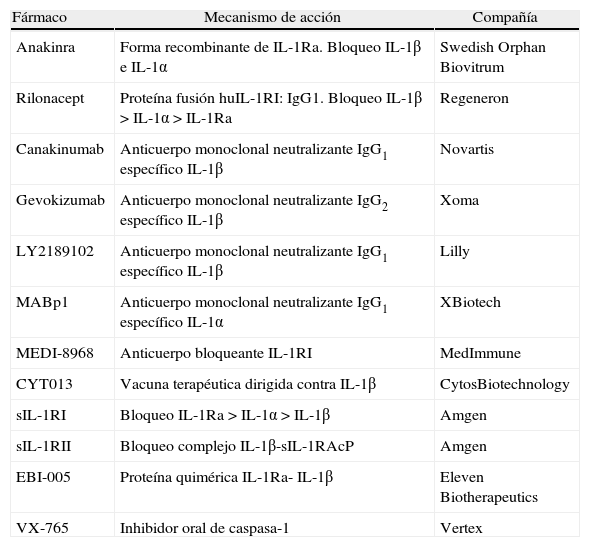
Los avances realizados en el conocimiento de las bases fisiopatológicas de las enfermedades autoinflamatorias hereditarias, y muy especialmente las evidencias obtenidas de la implicación de la vía de la IL-1β y el inflamasoma en las mismas, constituyen la base para la aplicación clínica de los agentes bloqueantes de IL-1. Durante los últimos años estos agentes se han empleado para el tratamiento de pacientes afectos de determinadas enfermedades autoinflamatorias, tanto hereditarias como no hereditarias, convirtiéndose en algunas ocasiones en sus tratamientos de primera línea. Actualmente, son 3 los agentes biológicos bloqueantes de la IL-1 aprobados por las agencias americana (FDA) y europea (EMA), mientras que hay todo un conjunto de fármacos en diferentes ensayos clínicos19 (tabla 4). A continuación, se exponen las características principales de cada uno de los agentes que cuentan con la aprobación de las agencias reguladoras.
Clasificación de los agentes bloqueantes de interleucina-1
| Fármaco | Mecanismo de acción | Compañía |
| Anakinra | Forma recombinante de IL-1Ra. Bloqueo IL-1β e IL-1α | Swedish Orphan Biovitrum |
| Rilonacept | Proteína fusión huIL-1RI: IgG1. Bloqueo IL-1β>IL-1α>IL-1Ra | Regeneron |
| Canakinumab | Anticuerpo monoclonal neutralizante IgG1 específico IL-1β | Novartis |
| Gevokizumab | Anticuerpo monoclonal neutralizante IgG2 específico IL-1β | Xoma |
| LY2189102 | Anticuerpo monoclonal neutralizante IgG1 específico IL-1β | Lilly |
| MABp1 | Anticuerpo monoclonal neutralizante IgG1 específico IL-1α | XBiotech |
| MEDI-8968 | Anticuerpo bloqueante IL-1RI | MedImmune |
| CYT013 | Vacuna terapéutica dirigida contra IL-1β | CytosBiotechnology |
| sIL-1RI | Bloqueo IL-1Ra>IL-1α>IL-1β | Amgen |
| sIL-1RII | Bloqueo complejo IL-1β-sIL-1RAcP | Amgen |
| EBI-005 | Proteína quimérica IL-1Ra- IL-1β | Eleven Biotherapeutics |
| VX-765 | Inhibidor oral de caspasa-1 | Vertex |
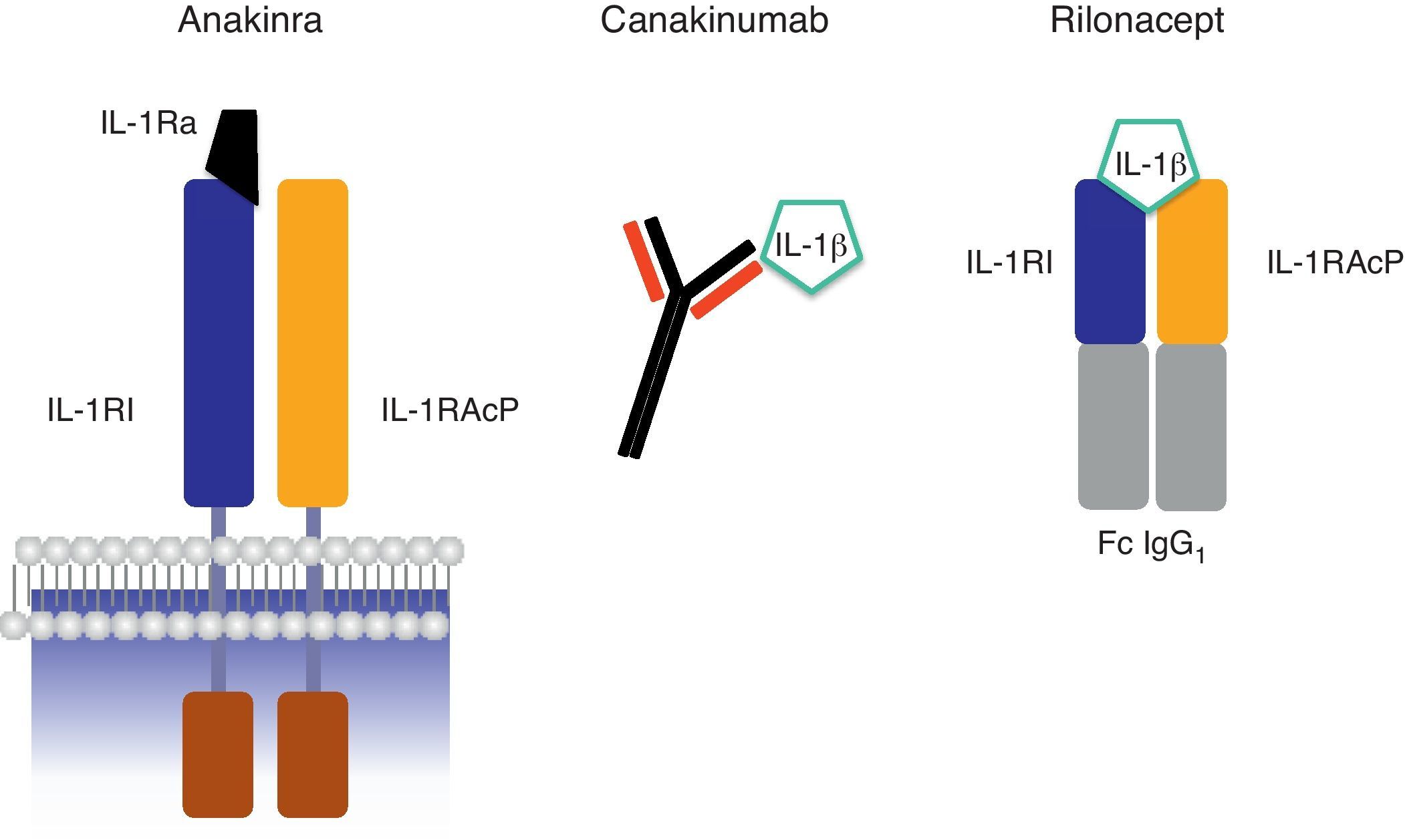
Se trata de la forma recombinante humana, no glicosilada, del antagonista del receptor de IL-1 (IL-1Ra). Se une al receptor i de la IL-1 impidiendo la unión de la proteína accesoria (IL-1RAcP) e inhibindo de manera competitiva a los 2 ligandos agonistas del receptor (IL-1α y IL-1β)4,19,26 (fig. 6). Presenta una vida media breve (4-6h), debiéndose administrar diariamente por vía subcutánea. Si bien las dosis estándar son 100mg/24h en adultos y 1mg/kg/24h en niños, estas pueden ser modificadas al alza o a la baja en función de la intensidad de la enfermedad inflamatoria. Entre los pocos efectos adversos observados el más frecuente es la aparición de dolor y/o hinchazón en el punto de inyección.

Esquema de los mecanismos de acción de los bloqueantes de IL-1 anakinra, canakinumab y rilonacept. Abreviaturas: IL-1β: interleucina-1β; IL-1RI: receptor de tipo i de la interleucina-1; IL-1RAcP: proteína accesoria del receptor de IL-1; IL-1Ra: antagonista del receptor de IL-1; Fc IgG1: fracción constante de la inmunoglobulina G1.
También conocida como IL-1 Trap, rilonacept es una proteína de fusión dimérica constituida por el fragmento Fc de una IgG1 asociada a los dominios extracelulares de unión al ligando del receptor i de la IL-1 (IL-1RI) y de la proteína accesoria del receptor de la IL-1 (IL-1RAcP)27. Debido a su estructura, rilonacept bloquea las acciones biológicas de las citocinas agonistas (IL-1α y IL-1β) y también la de la citocina antagonista (IL-1Ra) (fig. 6). Se administra por vía subcutánea semanalmente debido a que presenta una vida media ligeramente superior a los 8d.
Entre sus efectos adversos merecen destacarse: i) un aumento de la incidencia de infecciones leves y moderadas del tracto respiratorio superior y ii) la aparición de dolor y/o hinchazón en el punto de inyección. Asimismo, en los estudios publicados se demostró la aparición de anticuerpos dirigidos contra los fragmentos del receptor IL-1RI/IL-1RacP de rilonacept, si bien no ha quedado bien establecido si dichos anticuerpos originan o no una pérdida de la eficacia del tratamiento a largo plazo28,29.
Canakinumab (nombre comercial Ilaris™)Se trata de un anticuerpo monoclonal totalmente humanizado dirigido contra la citocina IL-1β que bloquea de manera selectiva las acciones de la citocina IL-1β pero preserva las acciones de las citocinas IL-1α e IL-1Ra30 (fig. 6). Se administra por vía subcutánea, al igual que los fármacos comentados previamente. Su prolongada vida media (≈28d) permite establecer una pauta de administración más cómoda, siendo la pauta estándar una dosis cada 8 semanas. Al igual que los fármacos anteriores, tanto la dosis como la pauta de administración pueden ser moduladas al alza o a la baja en función de la enfermedad inflamatoria a tratar. Entre sus efectos adversos destacan: i) un ligero incremento en la incidencia de infecciones, habitualmente leves y localizadas preferentemente en el tracto respiratorio superior, y ii) también reacciones locales en el punto de inyección. No se han identificado hasta el momento ni desarrollo de anticuerpos anti-canakinumab ni pérdida de la eficacia del tratamiento con su uso prolongado.
- 1.
La autoinflamación es un mecanismo fisiopatológico presente en múltiples enfermedades consecuencia de una disregulación de la respuesta inmune innata y caracterizada por una inflamación anormalmente aumentada.
- 2.
En la última década se han identificado las bases genéticas responsables de unas 15 enfermedades autoinflamatorias hereditarias diferentes que se transmiten con un patrón de herencia mendeliano clásico.
- 3.
Durante estos años se ha demostrado el papel central de la interleucina-1 (IL-1) y del inflamasoma en la fisiopatología de la autoinflamación.
- 4.
El inflamasoma es un complejo multiproteico citoplasmático que genera la forma activa de ciertas interleucinas proinflamatorias (IL-1, IL-18, IL-33).
- 5.
El desequilibrio entre las citocinas proinflamatorias y antiinflamatorias observado en las enfermedades autoinflamatorias es consecuencia de una hiperactividad, intrínseca o extrínseca, del inflamasoma.
- 6.
El efecto global del incremento de la acción biológica de la IL-1 en las enfermedades autoinflamatorias ha permitido tratar, con éxito, a pacientes afectos de estas enfermedades con agentes bloqueantes de la IL-1.