





El síndrome de Sjögren (SS) es una de las enfermedades autoinmunes (EAI) más frecuentes. Se define como una epitelitis linfocitaria de las glándulas exocrinas y del epitelio de múltiples órganos. El compromiso de las glándulas lagrimales y salivales se expresa clínicamente con xerostomía y xeroftalmia (síndrome seco o sicca syndrome)1.
Hasta un tercio de los pacientes tiene complicaciones extraglandulares, que en ocasiones pueden ser graves. Puede presentarse solo (SS primario) o asociado a otras enfermedades reumáticas (SS secundario), principalmente al lupus eritematoso sistémico (LES), a la artritis reumatoide (AR) o en el contexto de diversos síndromes de solapamiento1.
La hiperactividad de los linfocitos B es una característica primordial en el SS. Se expresa con una llamativa hipergammaglobulinemia y la producción de autoanticuerpos (AAc). Uno de sus rasgos distintivos es la alta prevalencia de AAc anti-SSA/Ro y anti-SSB/La; tanto es así que forma parte de los criterios diagnósticos del SS primario2.
Se han propuesto varias clasificaciones para el diagnóstico de SS, siendo la más empleada y aceptada la del 2002. Entre los criterios se incluyen síntomas subjetivos de sequedad oral y ocular, además de pruebas dirigidas a objetivar la disminución de las secreciones glandulares salivales y lagrimales responsables de los síntomas de sequedad, la histopatología de las glándulas salivales y la presencia de Ac anti-SSA/Ro y anti-SSB/La3. Recientemente, el American College of Rheumatology (ACR) ha propuesto una nueva clasificación4, en un intento de optimizar el diagnóstico y poder establecer subgrupos de pacientes con una cierta homogeneidad. En esta los criterios son: 1) presencia de anti-SSA/Ro y/o anti-SSB/La o factor reumatoide positivo más anticuerpos antinucleares (ANA) a título ≤ a 1:320; 2) biopsia de glándula salival menor con focos de sialoadenitis linfocitaria; y 3) queratoconjuntivitis seca con un score de tinción ocular (con verde de lisamina o fluoresceína) ≤ de 3. Para establecer el diagnóstico de SS se deben cumplir al menos 2 criterios. Tiene una sensibilidad y especificidad del 93% y 95%, respectivamente4. Respecto a la clasificación del año 2002 se eliminaron los criterios relativos a los síntomas subjetivos de sequedad ocular y oral, así como las pruebas funcionales o morfológicas de las glándulas salivales (con baja especificidad) y el test de Schirmer para medir la producción lagrimal. La nueva clasificación no distingue entre SS primario y secundario. Debido a que aún no hay consenso, el ACR y la Liga Europea Contra el Reumatismo (EULAR) han designado un grupo de trabajo para unificar criterios.
Autoanticuerpos en el síndrome de SjögrenLos ANA están presentes entre el 60% y el 80% de los pacientes con SS2,5, y el patrón de inmunofluorescencia habitual es el nuclear moteado fino y el citoplasmático.
Anticuerpos anti-SSA/Ro y anti-SSB/LaLas células epiteliales de las glándulas salivales parecen cumplir un rol en la iniciación y desarrollo de la respuesta inmune local en el SS, siendo capaces de intervenir en la exposición de los Ag SSA/Ro y SSB/La al sistema inmune mediante una apoptosis elevada y liberación de autoantígenos (AAg)2. Estos AAc son los únicos marcadores serológicos contemplados en la clasificación del ACR del año 20023. Sin embargo, la detección de ANA (a título≥1:320) más la presencia de factor reumatoide pueden ser considerados equivalentes a la presencia de anti-SSA/Ro y SSB/La según los nuevos criterios4.
La presencia de anti-SSA/Ro 52 y 60kDa coexiste en una alta proporción de pacientes con SS. Por orden de frecuencia se detectan Ac anti-SSA/Ro 52 (67%), anti-SSA/Ro 60 (52%) y anti-SSB/La (49%)2. El Ac anti-SSA/Ro puede aparecer en solitario, pero es muy raro que lo haga el anti-SSB/La, que casi siempre aparece junto al anti-SSA/Ro6. Sin embargo, se ha observado que la presencia del anti-SSB/La es el marcador inmunológico más específico4. La presencia de estos AAc se ha relacionado con una mayor duración de la enfermedad antes del diagnóstico, extensa infiltración linfocitaria de las glándulas salivales menores, disfunción glandular exocrina grave y aumento recurrente del tamaño parotídeo2. Asimismo, títulos elevados de anti-SSA/Ro y anti-SSB/La se han asociado con una alta prevalencia de afectación extraglandular, siendo la vasculitis la más alarmante2,7. Los niveles de estos AAc se mantienen estables durante el curso de la enfermedad6.
DermatomiositisLa dermatomiositis (DM) es otra EAI que se asocia a la presencia de AAc. Pertenece al grupo de las miopatías inflamatorias idiopáticas (MII), junto a la polimiositis (PM) y a la miositis por cuerpos de inclusión. Afecta al músculo esquelético, a la piel y, a veces, a otros órganos como el pulmón, el corazón y las articulaciones. Hay variantes clínicas como la DM clásica, que tiene compromiso muscular y cutáneo; la DM amiopática, donde hay daño cutáneo característico en ausencia de miositis clínica o analítica; DM hipomiopática, cuando las manifestaciones cutáneas se asocian a evidencia de miositis subclínica (elevación de enzimas musculares); DM postmiopática, cuando pacientes con DM clásica previa presentan recuperación de la miositis, pero persisten las alteraciones cutáneas; y DM sine dermatitis, en la que no hay enfermedad cutánea evidente pero la biopsia muscular demuestra características histológicas de DM8.
Ante la sospecha de una DM debe procederse a la determinación de ANA mediante IFI sobre células HEp-2, pero este no es un método sensible ni tampoco tiene un patrón de fluorescencia específico, aunque los más frecuentes son el nuclear moteado fino y el citoplasmático granular. Es por eso que en caso de sospecha de DM o cualquier MII, hay que investigar también la presencia de AAc específicos. En algunos laboratorios se procede de manera inversa, investigando primero la existencia de AAc específicos agrupando antígenos (Ag) en perfiles de inmunoblot para su detección, que tiene alta sensibilidad. En el caso de algunos AAc se puede confirmar su presencia mediante IFI sobre células HEp-2 porque presentan patrones específicos. Si hubiese dudas al respecto se emplea la técnica de inmunoprecipitación, que es el gold standard. Existen a día de hoy métodos de enzimoinmunoanálisis (ELISA) y quimioluminiscencia que permiten cuantificar algunas especificidades, como el anti-Jo1.
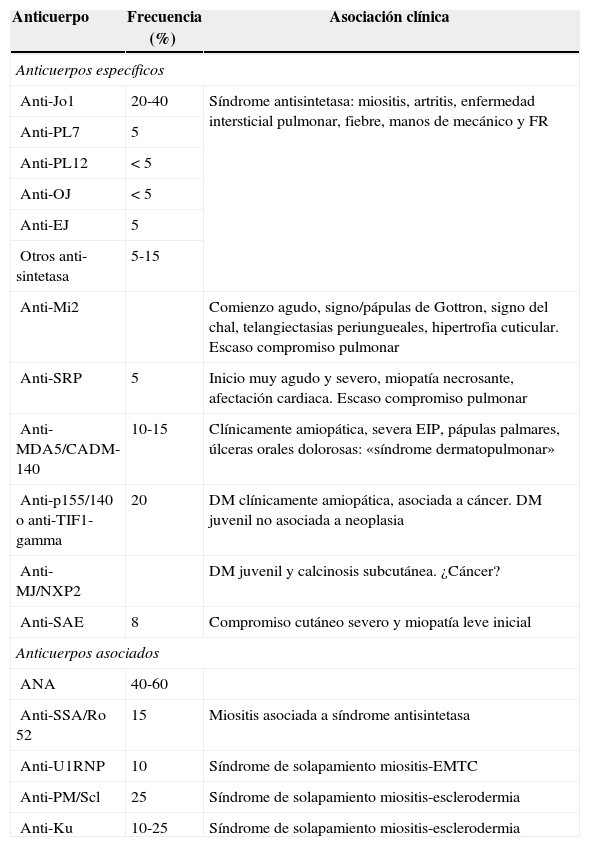
En pacientes con DM hay una fuerte asociación entre la presencia de determinados AAc y fenotipos clínicos diferentes. Se pueden encontrar 2 tipos de AAc. El primer grupo está representado por los AAc asociados a miositis (AAM) y suelen estar presentes en pacientes con otras EAI, como los síndromes de solapamiento. El segundo grupo corresponde a los AAc específicos de miositis (AEM), virtualmente ausentes en otras EAI y distrofias musculares1,8,9. Estos AEM son habitualmente excluyentes entre sí1. La presencia de un AAc no es un criterio diagnóstico de DM, pero su estudio es de utilidad para definir subgrupos clínicos más homogéneos y aportar valor pronóstico1,9,10. Aún no queda claro por qué surgen y si desempeñan un papel patológico en el proceso de la enfermedad, pero hay indicios que apoyan su relevancia fisiopatológica.
Anticuerpos específicos de miositisAnticuerpos anti-Jo1 y otros anticuerpos antisintetasaLas aminoacil-tRNA sintetasas son enzimas citoplasmáticas que catalizan la unión covalente de cada uno de los aminoácidos con su ARN de transferencia (tRNA) específico. Hay un único tRNA para cada aminoácido; así, por ejemplo, la histidil-tRNA sintetasa une la histidina a su tRNA. El complejo aminoácido-tRNA transfiere el aminoácido específico a la cadena polipeptídica de alargamiento de la manera en la que el ribosoma lee la secuencia que codifica un ARN mensajero (ARNm)11,12.
Debido a la localización citosólica de estas enzimas, la presencia de Ac antisintetasa dan un patrón citoplasmático en la inmunofluorescencia indirecta (IFI) sobre células HEp-2. La negatividad de la prueba no descarta la presencia de estos AAc1.
Los Ac contra la histidil-tRNA sintetasa (anti-Jo1) son los AEM más comunes11,12. Se identifican en un grupo de pacientes que presentan miositis, enfermedad intersticial pulmonar (EIP), artritis no erosiva, fiebre, fenómeno de Raynaud (FR) y lesiones hiperqueratósicas características en la cara radial y palmar de los dedos, conocidas como «manos de mecánico»11. Estos hallazgos clínicos se conocen con el nombre de síndrome antisintetasa. El anti-Jo1 se encuentra en el 25-30% de pacientes con miositis y, de estos el 60-70% tienen EIP11,12.
Hasta la fecha se han identificado AAc contra 7 tipos de aminoacil-tRNA sintetasas, además del anti-Jo1, a saber: anti-PL7 (treonil), anti-PL12 (alanil), anti-OJ (isoleucil), anti-EJ (glicil), anti-KS (asparraginil), anti-Zo (fenilalanil) y anti-Ha (tirosil)12. Cada uno de ellos tiene una prevalencia menor al 5% en miositis12. Entre el 90 y el 100% de los pacientes con Ac antisintetasa tienen EIP, mientras que entre uno y 2/3 de pacientes con miositis y enfermedad pulmonar tienen alguno de los Ac antisintetasas12. Es importante resaltar que hay enfermedad pulmonar en el 30% de los pacientes con miositis en ausencia de Ac antisintetasa, sugiriendo la posibilidad de que estos enfermos tengan AAc aún no identificados11.
Pacientes con anti-PL12 y otros Ac antisintetasa parecen tener mayor afección pulmonar sin enfermedad muscular clínicamente detectable. Al contrario, el 90% de los individuos con anti-Jo1 tienen miopatía y mayor frecuencia de artritis, fiebre y manos de mecánico comparado con pacientes con otros Ac antisintetasa11.
La evolución de la enfermedad está dominada por el compromiso pulmonar. El significado pronóstico de la EIP ha sido objeto de varios estudios. La tasa de supervivencia a 5 años en la última década ha subido hasta el 75-95%11, y se ha demostrado que el título de Ac anti-Jo1 se correlaciona (modestamente) con la actividad de la enfermedad y particularmente con el compromiso pulmonar11.
Anticuerpos anti-Mi-2El Ag Mi-2 es una helicasa nuclear con función reguladora de la transcripción cromosómica. Forma parte del complejo nucleosome-remodeling deacetylase (NuRD)1. Se ha demostrado que la expresión de Mi-2 es crucial para el correcto desarrollo y reparación de la membrana basal epidérmica9,11. En la IFI da un patrón de tinción nuclear moteado fino1.
Pacientes con DM y presencia de Ac anti-Mi-2 suelen presentar manifestaciones cutáneas llamativas, como el eritema heliotropo, la erupción en la parte dorsal superior y cervical (signo del chal) y la hipertrofia cuticular. Sin embargo, la presencia del anti-Mi-2 se asocia a un pronóstico favorable, con buena respuesta a esteroides y una baja incidencia de neoplasia asociada comparada con otros AAc9,11.
Hay estudios que demuestran la relación entre la latitud y la proporción relativa de DM entre pacientes con miositis. La radiación UV aumenta la expresión de Mi-2 en queratinocitos humanos. Al parecer, la radiación UV induce una dermatitis que deriva en mayor expresión de Mi-2, generándose una respuesta autoinmune contra este Ag, lo que explica por qué este AAc es más prevalente a menor latitud, donde la intensidad de la radiación UV es mayor11.
Asimismo, la expresión de Mi-2 en biopsias musculares de pacientes con DM es significativamente mayor que en las de pacientes con PM. Esto apoya la idea de que la expresión elevada de Mi-2 en tejidos dañados de individuos con DM conduce al daño por Ac anti-Mi-29.
Anticuerpos anti-signal recognition particleLa signal recognition particle (SRP) es un complejo citoplasmático que media la traslocación de polipéptidos a través del retículo endoplasmático1. El patrón de tinción que da la existencia de estos AAc en la IFI es citoplasmático difuso o granular1.
Los pacientes con DM y AAc anti-SRP se caracterizan por presentar una afectación muscular rápida y grave desde el inicio con debilidad progresiva, disfagia y niveles de creatincinasa elevados; inicialmente responden al tratamiento esteroideo, pero es habitual que se requieran otros inmunosupresores para el control de la enfermedad9,11. Las biopsias musculares revelan una miositis necrosante con escasa o nula inflamación9. Estos pacientes no suelen tener lesiones cutáneas, artritis ni EIP1,9,11. Los niveles de estos AAc se correlacionan con la actividad de la enfermedad y podrían ser útiles en el seguimiento evolutivo9.
Anticuerpos anti-MDA5/CADM 140La diana molecular de este AAc es una helicasa citoplasmática ARN-específica que funciona como un sensor que reconoce ARN virales. Esta proteína conduce a la expresión de genes antivirales y a la producción de interferón tipo i, dirigiendo la respuesta inmune intracelular para controlar la infección9,12. En la IFI estos AAc dan lugar a una tinción citoplasmática1.
Clínicamente se caracteriza por tener ausencia de afectación muscular y riesgo elevado de desarrollar EIP rápidamente progresiva y refractaria. La afectación mucocutánea consiste en úlceras cutáneas y orales, pápulas inflamatorias en palmas, pápulas de Gottron (fig. 1) y artritis/artralgias. Este importante compromiso cutáneo y pulmonar, con mínima o nula afectación muscular, se conoce como «síndrome dermato-pulmonar»9.
Anticuerpos anti-p155/140 o anti-TIF1-gamma, anti-SSA/Ro 52 a títulos elevados, pero estudio de AEM negativos.")
Los Ag de este AAc corresponden a un grupo de proteínas que pertenecen a la familia del factor intermediario de la transcripción 1 (transcription intermediary factor-1), que incluye las proteínas TIF1-gamma, alfa y beta, siendo la primera la diana más frecuente de este AAc9. En la IFI sobre células HEp-2 se corresponde con un patrón citoplasmático1.
La presencia de este AAc es útil para el diagnóstico de miositis asociada a cáncer. La utilidad radica en su alto valor predictivo negativo, con una baja probabilidad de neoplasia en pacientes con negatividad del Ac anti-p155/140 en pacientes con DM13.
La familia de las proteínas TIF1 está involucrada en la carcinogénesis y está sobreexpresada en diversos tumores. Los AAc contra estas proteínas podrían representar una respuesta antitumoral inicial12. También puede aparecer en formas juveniles de DM, pero en estos casos no se asocia a neoplasia1,11.
Aún no se dispone de kits comerciales específicos para su detección, por lo que actualmente su determinación se lleva a cabo únicamente en laboratorios de investigación mediante inmunoprecipitación.
Anticuerpos anti-MJ/NXP2Este AAc se dirige contra una proteína nuclear que está involucrada en la regulación de la proteína p53 inducida por el envejecimiento celular en respuesta a señales oncogénicas12. En la IFI expresa un patrón nuclear moteado fino con múltiples puntos nucleares14.
Se observa hasta en el 25% de DM juvenil en algunas series, se asocia a calcinosis subcutánea y contracturas musculares e indica gravedad. También puede encontrarse en formas del adulto con fenotipo similar. Se sugiere que podría ser un potencial marcador asociado a neoplasia, pero la evidencia es contradictoria10,12.
Anticuerpos anti-SAEEl Ag es una enzima llamada small ubiquitin-like modifier activating enzime (SUMO-1), una proteína nuclear que determina una conjugación enzimática postraslacional de proteínas diana. Da un patrón de tinción en la IFI nuclear moteado grueso sin tinción del nucléolo15. No se estudia de rutina, sino en centros de investigación.
Este AAc se encuentra en aproximadamente un 8% de los pacientes con DM. Se ha relacionado con lesiones cutáneas intensas acompañadas de compromiso muscular leve iniciales, pero la mayoría de los pacientes acaban desarrollando miositis y manifestaciones sistémicas llamativas en el curso de la enfermedad12.
Anticuerpos asociados a miositisAnticuerpos anti-SSA/Ro 52Este AAc se encuentra en más del 30% de los pacientes con miositis, frecuentemente asociado a Ac antisintetasa. La alta prevalencia del anti-SSA/Ro 52 en pacientes afectos de miopatía autoinmune y/o EIP y su asociación a AEM, fundamentalmente al anti-Jo1 (en alrededor de un 70% de los casos), sugiere que las partículas antigénicas SSA/Ro 52 podrían participar en el inicio o mantenimiento del daño muscular y pulmonar12. Aún no se conoce completamente la relevancia clínica del Ac anti-Ro 52 en individuos con miositis, pero hay evidencias que sugieren que podría tener valor pronóstico en pacientes con DM/PM y Ac anti-Jo1, habiéndose asociado con EIP, miositis y compromiso articular más grave comparado con pacientes con anti-Jo1 positivo pero sin anti-SSA/Ro 5212.
Cuando se considera en un paciente el diagnóstico de DM o más específicamente de síndrome antisintetasa, se sugiere determinar el anti-SSA/Ro 52 de forma separada16. En la tabla 1 se resumen los AAc en DM, tanto los asociados como los específicos.
Autoanticuerpos en la dermatomiositis y sus rasgos clínicos distintivos
| Anticuerpo | Frecuencia (%) | Asociación clínica |
|---|---|---|
| Anticuerpos específicos | ||
| Anti-Jo1 | 20-40 | Síndrome antisintetasa: miositis, artritis, enfermedad intersticial pulmonar, fiebre, manos de mecánico y FR |
| Anti-PL7 | 5 | |
| Anti-PL12 | <5 | |
| Anti-OJ | <5 | |
| Anti-EJ | 5 | |
| Otros anti-sintetasa | 5-15 | |
| Anti-Mi2 | Comienzo agudo, signo/pápulas de Gottron, signo del chal, telangiectasias periungueales, hipertrofia cuticular. Escaso compromiso pulmonar | |
| Anti-SRP | 5 | Inicio muy agudo y severo, miopatía necrosante, afectación cardiaca. Escaso compromiso pulmonar |
| Anti-MDA5/CADM-140 | 10-15 | Clínicamente amiopática, severa EIP, pápulas palmares, úlceras orales dolorosas: «síndrome dermatopulmonar» |
| Anti-p155/140 o anti-TIF1-gamma | 20 | DM clínicamente amiopática, asociada a cáncer. DM juvenil no asociada a neoplasia |
| Anti-MJ/NXP2 | DM juvenil y calcinosis subcutánea. ¿Cáncer? | |
| Anti-SAE | 8 | Compromiso cutáneo severo y miopatía leve inicial |
| Anticuerpos asociados | ||
| ANA | 40-60 | |
| Anti-SSA/Ro 52 | 15 | Miositis asociada a síndrome antisintetasa |
| Anti-U1RNP | 10 | Síndrome de solapamiento miositis-EMTC |
| Anti-PM/Scl | 25 | Síndrome de solapamiento miositis-esclerodermia |
| Anti-Ku | 10-25 | Síndrome de solapamiento miositis-esclerodermia |
La ES es una enfermedad multisistémica y heterogénea, en la que se producen 3 alteraciones fundamentalmente: vasculopatía de pequeño vaso, activación del sistema inmune con producción de AAc y disfunción fibrobástica que conlleva a un exceso de depósito de matriz extracelular17. En la piel esto se traduce en un engrosamiento y endurecimiento de la misma. Sucede lo mismo con otros órganos internos, como el pulmón, el esófago y el resto del tubo digestivo, el corazón y el riñón, entre otros. Las manifestaciones clínicas van a depender de la presencia y el grado de afectación cutánea y de los distintos órganos. Se ha observado en los pacientes con compromiso orgánico grave que este ocurre tempranamente en el curso de la enfermedad, y además puede aparecer incluso antes de que se cumplan los criterios de ES18.
La ES se divide en 4 subtipos, en función del tipo de afectación cutánea: 1) ES temprana o pre-ES, definida por la presencia de FR, alteraciones en la capilaroscopia digital y AAc específicos, pero sin engrosamiento de la piel; 2) ES cutánea limitada, en la que hay compromiso cutáneo distal sin sobrepasar los codos, las rodillas y la cara; 3) ES cutánea difusa, con afectación cutánea proximal a estas articulaciones; y 4) ES sin esclerodermia, dominada por las alteraciones orgánicas, sin afectar a la piel19.
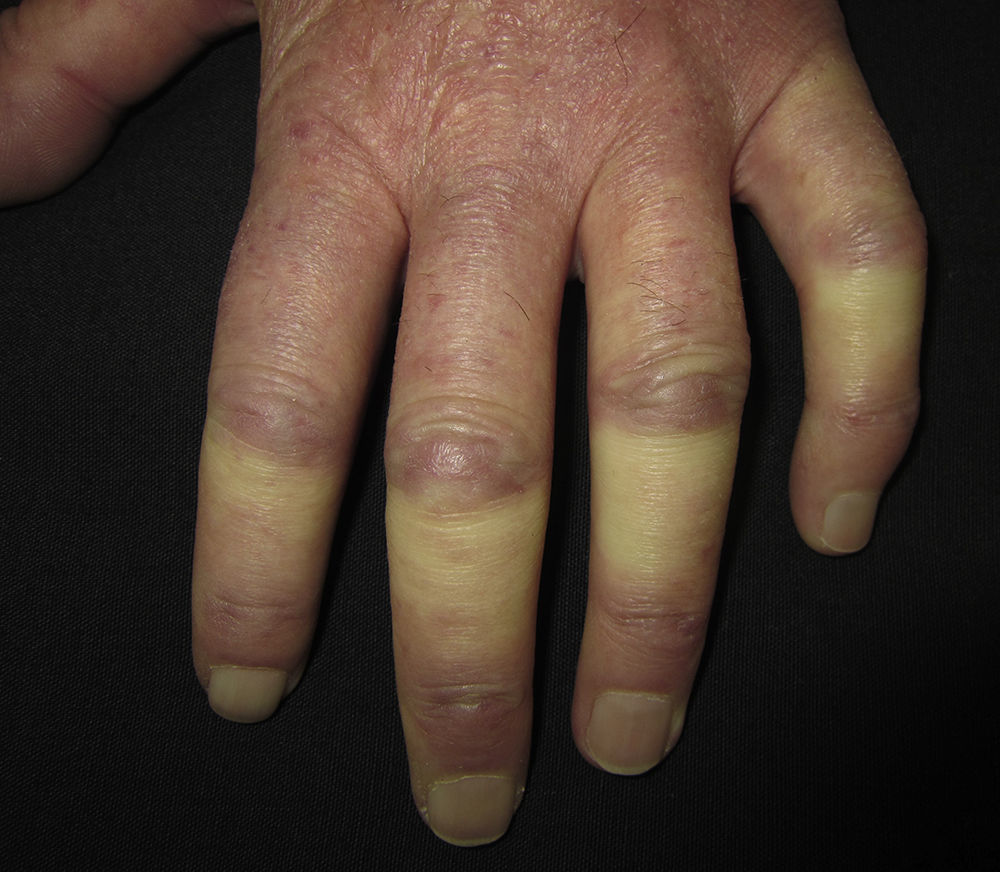
Con el objetivo de mejorar la clasificación de los pacientes con ES y disminuir el umbral para diagnosticar a estos pacientes en estadios muy precoces se ha propuesto recientemente un sistema de clasificación con puntuación que consta de 8 ítems, incluyendo: 1) engrosamiento cutáneo de los dedos de ambas manos que se extiende proximalmente a las articulaciones metacarpofalángicas (criterio suficiente para diagnosticar ES); 2) engrosamiento cutáneo o inflamación de los dedos, en forma de «dedos en salchicha» o esclerodactilia distal a las articulaciones metacarpofalángicas, pero proximal a la interfalángica proximal; 3) lesiones en el pulpejo de los dedos a modo de úlceras digitales o escaras puntiformes; 4) telangiectasias; 5) anomalías en los capilares de la matriz ungueal; 6) afectación pulmonar (hipertensión pulmonar o EIP); 7) FR; y 8) AAc específicos (anti-centrómero, anti-topoisomerasa i o anti-ARN polimerasa iii [anti-ARNP iii])17. Estos criterios no son aplicables a pacientes que padezcan síndromes esclerodermiformes de etiologías conocidas. Esta clasificación es útil para incluir pacientes en estudios de ES, pero no está validado su empleo en la práctica clínica diaria17. De manera similar se ha utilizado el concepto de «bandera roja» (‘red flag’) para definir la tríada de FR, ANA positivos y «dedos en salchicha», considerado este último un signo muy precoz de ES18 (fig. 2). Se ha observado que casi el 90% de los pacientes con esta tríada «bandera roja» presentaron además Ac anti-centrómero, anti-topoisomerasa i y/o alteraciones en la capilaroscopia, lo que ya los clasificaría como ES temprana18. Incluso en ausencia de ANA, la presencia de «dedos en salchicha» podría ser un signo de alarma en pacientes con FR, ya que se ha observado que pueden progresar a ES18. Además de los factores predictivos bien caracterizados, los «dedos en salchicha» constituyen un importante signo de sospecha para ES en estadio muy precoz en pacientes con FR18.

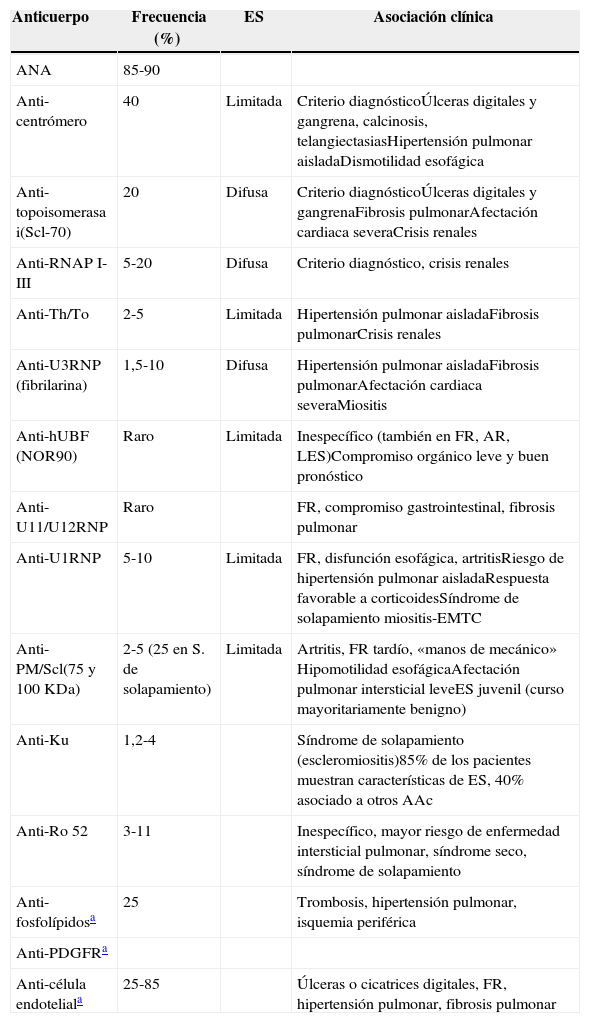
En la ES los ANA por IFI presentan una sensibilidad entre el 85% y 90%, con una especificidad algo superior al 50%1,20,21. Uno de los principales pilares para su clasificación es la presencia de AAc específicos. Sin embargo, la ausencia de AAc no descarta el diagnóstico de ES. Los ANA que se asocian a la forma «pura» de la enfermedad son 5: anti-centrómero, anti-topoisomerasa i (anti-Scl70), anti-RNAP I-III, anti-Th/To y anti-U3RNP (fibrilarina). Es excepcional la coexistencia de 2 o más AAc en el mismo paciente y la positividad suele mantenerse durante el curso de toda la enfermedad1. Estos AAc se asocian a características demográficas, clínicas, orgánicas y de supervivencia muy específicas, por lo que su determinación puede ser útil en el pronóstico y la evolución de los pacientes con ES. Además, se han observado diferencias en frecuencia y gravedad del compromiso orgánico cuando se agrupa a los pacientes según el AAc presente, sugiriendo que los AAc específicos serían mejores indicadores del patrón clínico de ES que la afectación cutánea22,23.
Al iniciar el estudio de un paciente con características clínicas que sugieran una ES se debe practicar la IFI en un primer paso, como se ha detallado en apartados anteriores. Si resultara positiva sería conveniente realizar las pruebas serológicas pertinentes para identificar el AAc específico. Este último paso se puede obviar si el patrón observado en la IFI fuese el centromérico, ya que tiene una especificidad cercana al 100%. Los AAc asociados a la ES se resumen en la tabla 2, comentando brevemente los más relevantes.
Autoanticuerpos en la esclerosis sistémica
| Anticuerpo | Frecuencia (%) | ES | Asociación clínica |
|---|---|---|---|
| ANA | 85-90 | ||
| Anti-centrómero | 40 | Limitada | Criterio diagnósticoÚlceras digitales y gangrena, calcinosis, telangiectasiasHipertensión pulmonar aisladaDismotilidad esofágica |
| Anti-topoisomerasa i(Scl-70) | 20 | Difusa | Criterio diagnósticoÚlceras digitales y gangrenaFibrosis pulmonarAfectación cardiaca severaCrisis renales |
| Anti-RNAP I-III | 5-20 | Difusa | Criterio diagnóstico, crisis renales |
| Anti-Th/To | 2-5 | Limitada | Hipertensión pulmonar aisladaFibrosis pulmonarCrisis renales |
| Anti-U3RNP (fibrilarina) | 1,5-10 | Difusa | Hipertensión pulmonar aisladaFibrosis pulmonarAfectación cardiaca severaMiositis |
| Anti-hUBF (NOR90) | Raro | Limitada | Inespecífico (también en FR, AR, LES)Compromiso orgánico leve y buen pronóstico |
| Anti-U11/U12RNP | Raro | FR, compromiso gastrointestinal, fibrosis pulmonar | |
| Anti-U1RNP | 5-10 | Limitada | FR, disfunción esofágica, artritisRiesgo de hipertensión pulmonar aisladaRespuesta favorable a corticoidesSíndrome de solapamiento miositis-EMTC |
| Anti-PM/Scl(75 y 100 KDa) | 2-5 (25 en S. de solapamiento) | Limitada | Artritis, FR tardío, «manos de mecánico» Hipomotilidad esofágicaAfectación pulmonar intersticial leveES juvenil (curso mayoritariamente benigno) |
| Anti-Ku | 1,2-4 | Síndrome de solapamiento (escleromiositis)85% de los pacientes muestran características de ES, 40% asociado a otros AAc | |
| Anti-Ro 52 | 3-11 | Inespecífico, mayor riesgo de enfermedad intersticial pulmonar, síndrome seco, síndrome de solapamiento | |
| Anti-fosfolípidosa | 25 | Trombosis, hipertensión pulmonar, isquemia periférica | |
| Anti-PDGFRa | |||
| Anti-célula endoteliala | 25-85 | Úlceras o cicatrices digitales, FR, hipertensión pulmonar, fibrosis pulmonar |
AAc: autoanticuerpos; AR: artritis reumatoide; EMTC: enfermedad mixta del tejido conectivo; ES: esclerosis sistémica; FR: fenómeno de Raynaud; LES: lupus eritematoso sistémico; PDGFR: platelet derived growth factor receptor.
Presentes en el 50% de los pacientes con ES. Como su nombre indica, en la IFI sobre células HEp-2 se tiñen los centrómeros. Las proteínas centroméricas están localizadas en la parte interna y externa del cinetocoro y se unen al aparato mitótico durante la mitosis. Los AAc anti-centrómero reconocen 4 proteínas, frecuentemente la CENP-B de 80KDa (95%) y en menor medida la CENP-A de 17KDa. La CENP-C y D son Ag excepcionales. El patrón centromérico no se visualiza si la diana es la CENP-F1,24.
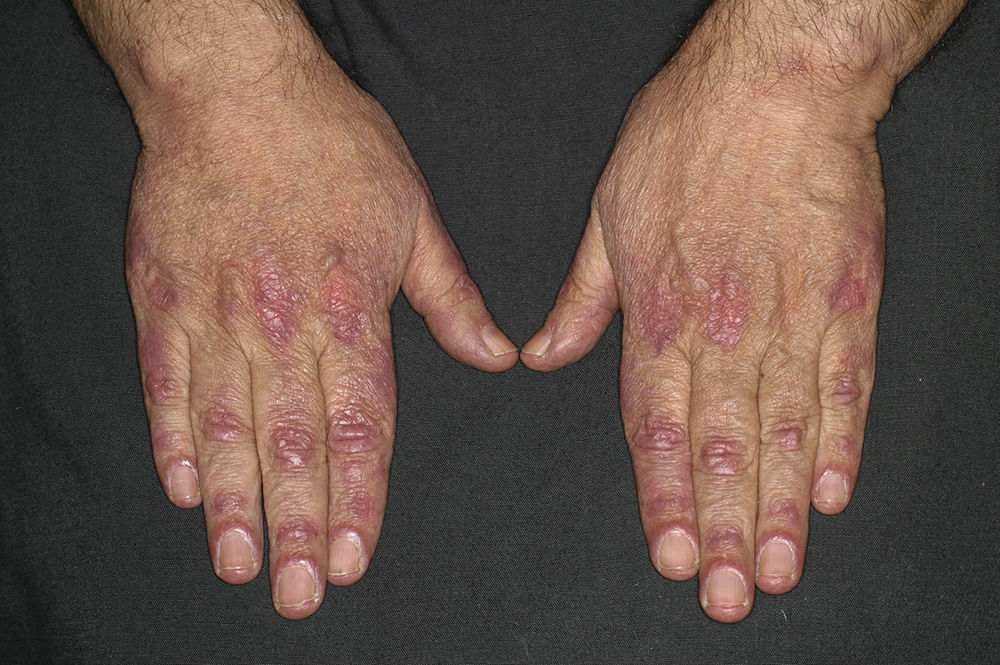
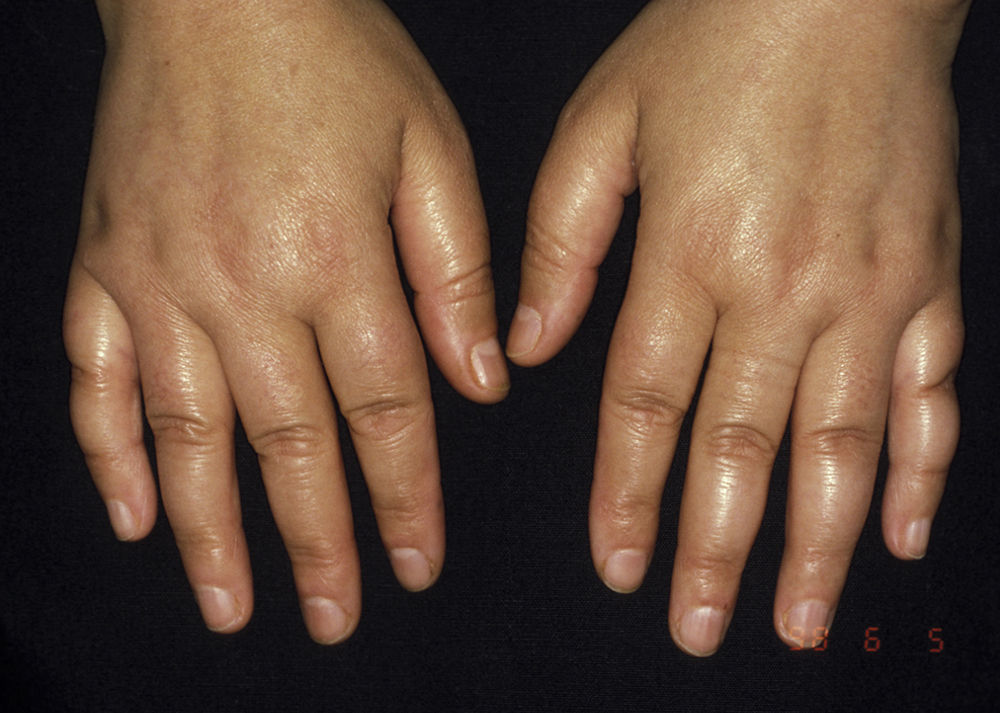
Este AAc está asociado a la afectación cutánea limitada19,20 (antiguamente denominada con el acrónimo CREST, de calcinosis, FR, alteraciones esofágicas, esclerodactilia y telangiectasias) (fig. 3), daño vascular periférico en forma de úlceras digitales y gangrena y calcinosis1. Sin embargo, en ocasiones puede observarse su positividad en la forma difusa (4,8%) y en la ES sin esclerodermia (7,6%)18. Las manifestaciones orgánicas más importantes son la hipomotilidad esofágica y la hipertensión pulmonar aislada. Se puede presentar hasta en el 30% de los pacientes con cirrosis biliar primaria, de los cuales la mitad pueden presentar signos de ES cutánea limitada. La mortalidad en este grupo de pacientes es menor que en el grupo con AAc anti-topoisomerasa i. Los Ac anti-centrómero pueden presentarse años antes del inicio de los síntomas y los títulos suelen ser estables a lo largo de la enfermedad, sin relacionarse con la gravedad1,20,22.

Manos de un paciente con esclerosis sistémica cutánea limitada y anticuerpos anticentrómero. Nótese la desaparición de las líneas de la piel en la cara dorsal y la ausencia de anejos debido a la fibrosis. En la cara palmar se observan telangiectasias, úlceras puntiformes en los pulpejos de los dedos, amputaciones distales y pterigium ungueal inverso.
El Ag Scl70 es nuclear y es un producto de la degradación de la topoisomerasa i funcionalmente activo. La función de esta enzima es catalizar el superenrollamiento del ADN previo a la acción de la ADN-polimerasa. Así, corta el ADN de doble cadena formando uno de cadena simple transitoria y permite que se desenrolle24. En la IFI la presencia de este AAc suele dar un patrón mixto con tinción de núcleo, nucléolos y cromosomas en metafase25, por lo que a menudo se describe como homogéneo con nucleolar. Aunque es un patrón característico, no es específico de Scl70, por lo que siempre es necesaria la confirmación con otros métodos como ELISA o inmunoblot.
Este AAc se encuentra en el 20-40% de pacientes con ES. Un 50-70% de los individuos con afectación cutánea difusa y el 25% de los que padecen la forma cutánea limitada tienen este Ac. Las variaciones en la frecuencia dependen de las diferencias étnicas1,20. Es indicativo de mal pronóstico: parece asociarse con enfermedad más grave y afectación difusa de la piel y multisistémica, específicamente con EIP de inicio temprano y compromiso cardiaco. Asimismo, su presencia puede predecir el desarrollo de crisis renales22. En pacientes con FR sugiere la posibilidad de evolucionar hacia una ES y, al igual que los Ac anti-centrómero, pueden preceder en años el inicio de los síntomas1. Su asociación con cáncer es controvertida20. Algunos estudios apoyan que la negativización de Ac anti-topoisomerasa i conlleva la remisión de la enfermedad1,20.
Anticuerpos anti-ARN polimerasa i, ii y iiiEl complejo de la ARN polimerasa (ARNP) se localiza en el nucléolo y transcribe genes de moléculas precursoras del ARN ribosómico. La II y la iii se sitúan en el nucleoplasma24. En el estudio de IFI se puede observar un patrón nuclear moteado fino con nucleolar.
Los Ac anti-RNAP I y III suelen coexistir y juntos son altamente específicos de ES1. Aparecen en el 5-20% de los pacientes afectos de ES. Se correlacionan con compromiso cutáneo difuso y crisis renales, no así con la fibrosis pulmonar, que en este grupo de pacientes es infrecuente. Por esto mismo y por la buena respuesta terapéutica a los inhibidores de la enzima convetidora de la angiotensina (IECA) en el manejo de la enfermedad renal, la supervivencia es mayor que la de los pacientes con AAc frente a la topoisomerasa i y U3RNP. El anti-RNAP II es inespecífico1,20.
Anticuerpos anti-Th/ToEstos AAc reconocen una proteína común a 2 de las ribonucleoproteínas pequeñas, la ARN-asa MRP y la ARN-asa P, que son endorribonucleasas24. Dan un patrón de tinción nucleolar en la IFI sobre células HEp-2. Son específicos, pero poco frecuentes en pacientes con ES (2-5%) y son marcadores de mal pronóstico. Se detectan en pacientes con enfermedad cutánea limitada, pero con compromiso sistémico extenso, con fibrosis pulmonar grave, hipertensión pulmonar secundaria y crisis renales. Ocasionalmente también pueden detectarse en pacientes con FR aislado1,20.
Anticuerpos anti-U3-ribonucleoproteico (anti-fibrilarina)Su antígeno es la fibrilarina, una proteína básica de 34 KDa, componente del complejo nucleolar U3-ribonucleoproteico. Al igual que la mayoría de los AAc en ES, da un patrón de tinción nucleolar en la IFI. Se encuentran en el 1,5-10% de los individuos con ES y son muy específicos de esta enfermedad, aunque excepcionalmente también se han detectado en LES20.
Su significación clínica depende de la etnia. Así, por ejemplo, los caucásicos, los afroamericanos y los japoneses desarrollan un compromiso cutáneo extenso, con vasculopatía periférica (úlceras puntiformes y gangrena), mientras que los individuos afroamericanos tienen además mayor compromiso sistémico, dominado por fibrosis pulmonar, hipertensión pulmonar y crisis renales, que empeoran el pronóstico20. En pacientes con FR estos AAc pueden ser indicativos de un futuro desarrollo de ES1.
Otros AAc que no son ANA que parecen tener un papel patogénico, con sensibilidad y especificidad elevadas en ES, son los anti-célula endotelial, los anti-fosfolípidos y los anti-receptor del factor de crecimiento derivado de las plaquetas (PDGFR). Los 2 primeros parecen estar más relacionados a los fenómenos vasculares de la ES1,20.
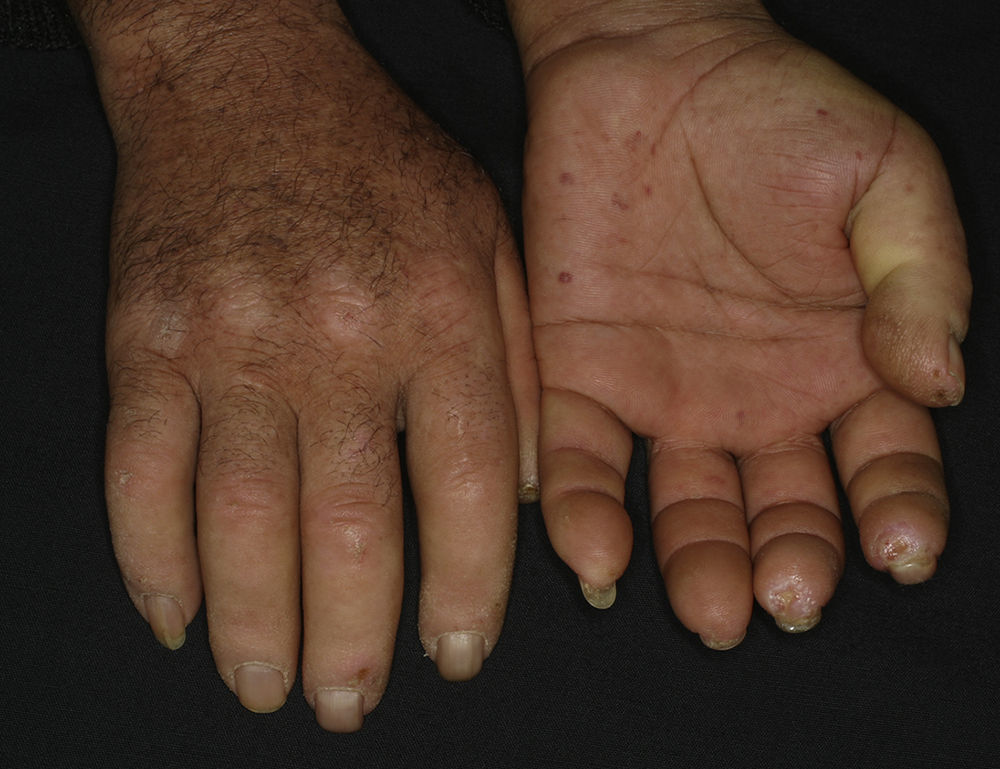
Enfermedad mixta del tejido conectivoLa enfermedad mixta del tejido conectivo (EMTC) o síndrome de Sharp es una enfermedad sistémica autoinmune que se caracteriza por presentar manifestaciones clínicas sugestivas de LES, ES y MII superpuestas, como el FR, dactilitis, poliartritis, enfermedad pulmonar y dismotilidad esofágica, entre otras (fig. 4). Es típica la presencia del Ac anti-U1RNP, generalmente a títulos elevados, aunque no es específico, ya que se puede encontrar en otras EAI26.

Hay 3 clasificaciones diagnósticas vigentes a día de hoy. La de Sharp et al., que fueron quienes describieron la enfermedad, es la que menor sensibilidad diagnóstica tiene (41%). Las clasificaciones de Kasukawa y de Alarcón-Segovia tienen sensibilidades similares, en torno al 75%. En las 3 se requiere la presencia del anti-U1RNP para el diagnóstico de la enfermedad27. Sin embargo, la presencia de títulos bajos de anti-U1RNP haría más plausible el diagnóstico de otra EAI, dado que son inespecíficos. Los Ac anti-dsADN, anti-Sm y anti-SSA/Ro pueden aparecer transitoriamente en individuos afectos de EMTC, pero si resultaran dominantes o persistentes hay mayor probabilidad de que el paciente padezca una enfermedad distinta a la EMTC1.
En un estudio de Szodoray et al. publicado recientemente se han perfilado 3 subgrupos dentro de la EMTC, en función de manifestaciones clínicas comunes y de marcadores serológicos concretos28. El primero está representado por pacientes que presentan predominantemente compromiso vascular, como el FR, livedo reticularis, dactilitis, trombosis vascular e hipertensión pulmonar. Este subgrupo tiene mayor prevalencia de ciertos AAc, como los anti-fosfolípidos y los anti-célula endotelial. Asimismo, observaron que el pronóstico en estos pacientes era peor y estaba condicionado por la afectación pulmonar. El segundo subgrupo presenta más frecuentemente EIP, dismotilidad esofágica y miositis. En este no detectaron un AAc relevante estadísticamente significativo, pero sí encontraron hallazgos patológicos mediados por inmunocomplejos en las biopsias estudiadas. El tercer subgrupo está representado por los pacientes que exhiben artritis erosiva, deformidad articular y AAc anti-péptido citrulinado.
Autoanticuerpos en la enfermedad mixta del tejido conectivoAnticuerpos anti-U1RNPEl Ag es un subtipo de ribonucleoproteínas que forman parte del espliceosoma, estructura proteica compleja situada en el citoplasma, que es el sitio donde el pre-ARN se convierte en ARNm maduro. Los Ac anti-U1RNP identifican las porciones antigénicas de 70KDa, A y C29. El anti-Sm también se dirige contra el espliceosoma, pero reconoce otras fracciones proteicas. En el LES es frecuente encontrarlo junto al anti-Sm, pero en la EMTC suele estar solo24. La existencia del Ac anti-U1RNP exhibe un patrón nuclear moteado grueso en la IFI sobre células HEp-2.
Síndromes de solapamientoLos síndromes de solapamiento (en inglés, overlap syndromes) se definen como enfermedades que satisfacen criterios diagnósticos de 2 o más EAI en el mismo o en diferentes momentos30. Se ha descrito una gran cantidad de síndromes de solapamiento asociados a muchos AAc no específicos, excepto los antisintetasas, los anti-U1RNP (ya descritos en apartados anteriores), el anti-PM/Scl y el anti-Ku. En la tabla 3 se expone la clasificación.
Clasificación del síndrome de solapamiento
| Asociado a autoanticuerpos específicos |
| EMTC |
| Síndrome antisintetasa |
| Esclero(dermato)miositis |
| LES-síndrome de Sjögren |
| No asociado a autoanticuerpos específicos |
| Rupus (síndrome de solapamiento de AR y LES) |
| ES-SS |
| ES-AR |
| LES-ES |
| AR-SS |
| PM-SS |
| Anti-U1RNP |
| Antisintetasas (Jo-1, PL7, PL12, etc.) |
| Anti-PM/Scl |
| Anti-SSB/La |
AR: artritis reumatoide; EMTC: enfermedad mixta del tejido conectivo; ES: esclerosis sistémica; LES: lupus eritematoso sistémico; PM: polimiositis; SS: síndrome de Sjögren.
Adaptada de Iaccarino et al.30.
El Ag está localizado en el sitio de ensamblaje del ribosoma en el nucléolo y forma parte del exosoma humano, estructura proteica compleja involucrada en el procesamiento del ARN durante la síntesis de proteínas29. Dos subunidades de esta estructura, de 75 y 100KDa, son los principales componentes antigénicos20. Da un patrón nucleolar homogéneo en la IFI.
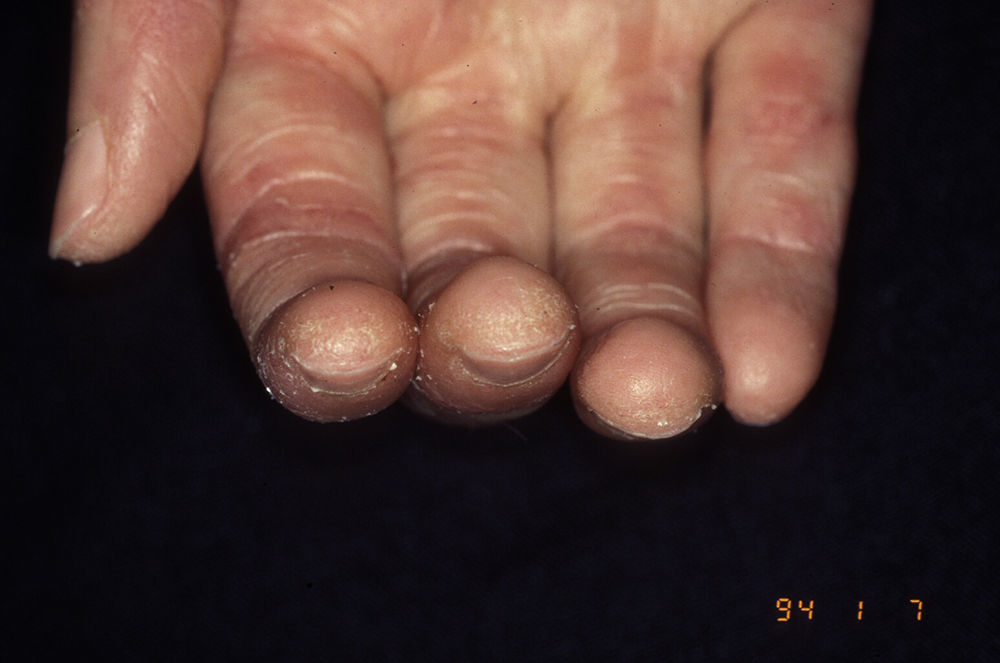
Es marcador de la escleromiositis, el solapamiento de ES limitada y (dermato)miositis. Un tercio de pacientes con escleromiositis presentan este AAc. El FR es característico y suele aparecer tardíamente en el curso de la enfermedad, se acompaña de artritis y de «manos de mecánico» (fig. 5). Inicialmente se relacionó con buen pronóstico y buena respuesta a corticoides, pero no son excepcionales los casos con enfermedad sistémica grave20. La esclerosis juvenil con anti-PM/Scl es la ES más común en la edad pediátrica, con un curso mayoritariamente benigno1.
Anticuerpos anti-Ku
Este AAc se detecta en un 2-25% de los pacientes con escleromiositis1,20. Da un patrón nuclear moteado fino con tinción de nucléolos en la IFI y el Ac se dirige contra un heterodímero nuclear. No tiene implicación pronóstica.
Enfermedad indiferenciada del tejido conectivoExisten pacientes con manifestaciones clínicas y serológicas típicas de las enfermedades autoinmunes, pero que no cumplen criterios suficientes para ser clasificados como una de ellas31. En un estudio longitudinal de pacientes con manifestaciones iniciales de enfermedad indiferenciada del tejido conectivo (EITC) se observó que al cabo de 5 años la mayoría (65%) mantenía esta condición (EITC «estable»), un tercio evolucionó hacía una EAI bien definida (EITC «incompleta») y una minoría se curaron32.
ConclusionesEntonces, ¿qué, cómo y cuándo solicitar un estudio serológico de autoinmunidad?Se recomienda solicitar ANA por IFI como prueba de primer nivel ante un paciente con síntomas y signos sugestivos de EAI. Es decir, cuando hay una sospecha clínica elevada. Si los ANA son positivos a título igual o superior a 1:160, el segundo paso sería solicitar la identificación de los Ac anti-ENA pertinentes según la sospecha clínica y el patrón de la IFI. La manera alternativa de hacerlo es pactando previamente con el personal de laboratorio los pasos a seguir en caso de resultar la IFI positiva. Esto permitiría ahorrar recursos humanos y materiales, además de abreviar tiempos en el diagnóstico en beneficio del paciente.
Un escenario posible, aunque menos frecuente, es la obtención de una prueba de ANA negativa en un paciente con alta sospecha clínica de padecer una EAI. Lo primero es confirmar el tipo de prueba utilizada. Si se tratara de un ELISA es imperativo repetir la prueba mediante IFI, que es la prueba de referencia. De haber sido la IFI negativa se justificaría solicitar Ac anti-ENA, dado que hay algunas enfermedades, como las miositis autoinmunes, en las que la sensibilidad de los ANA es relativamente baja. En este último caso el algoritmo diagnóstico podría iniciarse directamente con una prueba de detección de Ag específicos (por ejemplo inmunoblot) y si resultara positiva, confirmar su presencia mediante IFI buscando el patrón específico del Ac positivo. Si esta última no pusiera de manifiesto el patrón esperado, o fuese negativa, se podría hacer la prueba de referencia que es la inmunoprecipitación.
En el tema que nos ocupa es fundamental el trabajo conjunto de los especialistas clínicos (internistas, reumatólogos, dermatólogos, neumólogos, etc.) y de laboratorio, unos proporcionando la información clínica necesaria para poder interpretar correctamente un resultado positivo de la prueba solicitada, y eventualmente ampliar estudios con serologías complementarias si procede, y otros proporcionando datos acerca de las pruebas utilizadas y el algoritmo seguido durante el estudio serológico. Por otro lado, es importante que el médico clínico esté familiarizado con las características de cada prueba, así como el algoritmo utilizado en el laboratorio con el que trabaja, ya que cada institución maneja algoritmos adaptados a sus necesidades.
- •
Los Ac anti-SSA/Ro y anti-SSB/La son característicos del SS, aunque no son específicos de esta condición. En los criterios diagnósticos revisados recientemente se considera que la detección concomitante de ANA y factor reumatoide sería un criterio diagnóstico en casos sin anticuerpos anti-SSA/Ro y anti-SSB/La.
- •
En dermatomiositis, la IFI convencional para detectar ANA es poco sensible para estudiar si hay anticuerpos antisintetasa, por lo que se deben solicitar explícitamente los autoanticuerpos relacionados con esta enfermedad.
- •
En pacientes con dermatomiositis se pueden detectar 2 tipos de autoanticuerpos: los específicos (AEM) y los asociados (AAM). Los primeros son excluyentes entre sí, y se han relacionado con determinadas características de la enfermedad.
- •
En la esclerosis sistémica la sensibilidad de la IFI para detectar ANA es elevada, por lo que se debe solicitar como prueba de primer nivel ante la sospecha clínica; en un segundo paso, según el patrón de fluorescencia, se pueden solicitar pruebas específicas para desenmascarar cuál es el autoanticuerpo.
- •
Los autoanticuerpos en la esclerosis sistémica tienen importancia pronóstica, dado que se asocian a formas clínicas definidas, tanto de afectación cutánea como de compromiso orgánico. Estos son: anti-centrómero, anti-topoisomerasa i (Scl-70), anti-ARN polimerasa i y iii, anti-Th/To y anti-U3RNP (anti-fibrilarina).
- •
En pacientes con FR y ANA positivos la presencia de los «dedos en salchicha», tríada llamada «bandera roja», debe hacer sospechar una ES inicial.
- •
En la EMTC hay manifestaciones clínicas de LES, DM y ES; es característica la presencia de anticuerpos anti-U1RNP a títulos elevados.
- •
Hay múltiples síndromes de solapamiento descritos y suelen estar asociados a AAc no específicos. Uno de los sindromes más definidos es la escleromiositis, con presencia de anticuerpos anti-PM/Scl.
Los autores declaran no tener ningún conflicto de intereses.