


La pubertad es el periodo de transición entre la infancia y la edad adulta. Durante este periodo, se producen modificaciones que afectan a todos los órganos y las estructuras del cuerpo. Los cambios más evidentes incluyen:
- •
Adquisición de la talla adulta.
- •
Desarrollo de los caracteres sexuales secundarios.
- •
Maduración completa de las gónadas y las glándulas suprarrenales.
- •
Desarrollo completo de todos los órganos corporales.
Todas esas modificaciones aparecen por la activación del eje hipotálamo-hipofisario. Al disminuir la inhibición del sistema nervioso central (SNC) sobre el hipotálamo y la sensibilidad del gonadostato a los estrógenos circulantes, se inicia una secreción pulsátil del factor liberador de gonadotropinas (GnRH) que induce en la hipófisis un aumento de amplitud y frecuencia de los pulsos de LH. Este hecho se traduce en una estimulación gonadal con secreción de estradiol que provoca la aparición de los caracteres sexuales secundarios y, más adelante, la primera regla (menarquia).
Los cambios puberales fueron descritos por Tanner en 5 estadios referidos al desarrollo mamario (telarquia) y al vello púbico (pubarquia). En general, el primer signo de pubertad es la aceleración de crecimiento, seguido de la aparición del botón mamario entre los 9-11 años de edad cronológica. La aparición del vello púbico suele ser posterior a la telarquia, pero puede precederla en un 20% de los casos.
El estirón puberal en la niña se produce entre los estadios ii y iii de Tanner, alcanzándose el pico máximo de crecimiento en los 6-12 meses previos a la menarquia. La menarquia es un suceso tardío en el contexto del desarrollo puberal y se mantiene, desde hace varias décadas, entre los 12,5-12,8 años de edad cronológica.
Pubertad precozDefiniciónAparición de caracteres sexuales secundarios en las niñas, con tendencia a la progresión, antes de los 8 años. Estadísticamente, se define cuando los caracteres sexuales aparecen 2,5-3 desviaciones estándar por debajo de la edad promedio del inicio puberal. Tomando como referencia los estadios de Tanner, se incluye a toda niña que a los 8 años presente un estadio ii.
DiagnósticoAnamnesis familiar:
- •
Antecedentes familiares.
- •
Endocrinopatías.
- •
Talla de los progenitores.
Anamnesis personal:
- •
Enfermedades previas o actuales.
- •
Curva de crecimiento.
- •
Tratamientos farmacológicos.
- •
Fecha de inicio del desarrollo de los caracteres sexuales.
Exploración física:
- •
Talla y peso.
- •
Valoración del estadio puberal: mama, axila y genitales externos.
- •
Exploración general: fibromas en piel, manchas café con leche, hirsutismo, aumento del tamaño tiroideo.
Estudio analítico:
- •
TSH, FSH, LH, estradiol, test de GnRH.
- •
Si presenta signos de hirsutismo: testosterona, androstendiona, cortisol, 17OH-progesterona, sulfato de dehidroepiandrosterona.
Técnicas de imagen:
- •
Radiografía de la mano izquierda para estimación de la edad ósea.
- •
Ecografía ginecológica: valoración de relación de cuerpo/cérvix y características de los ovarios.
- •
Resonancia magnética craneal: descartar una causa de origen central.
- •
Fondo de ojo y valoración del campo visual, si la clínica lo indica.
Pubertad precoz central: son debidas a una activación del eje hipotálamo-hipófisis y por tanto son GnRH dependiente. Sus causas más frecuentes son:
- •
Idiopática: es la más frecuente en niñas. Máxima incidencia entre los 6 y los 8 años.
- •
Tumores intracraneales: generalmente antes de los 8 años; 10% en niñas.
- ∘
Hamartomas: son los más frecuentes.
- ∘
Disgerminomas.
- ∘
Craneofaringiomas.
- ∘
Astrocitomas.
- ∘
Pinealomas/gliomas.
- ∘
- •
Infecciosas: meningitis/encefalitis.
- •
Infiltrativas.
- •
Traumáticas.
- •
Malformaciones del SNC: hidro y macrocefalia.
- •
Hipotiroidismo de larga evolución.
- •
Irradiación del SNC.
- •
Sensibilización secundaria del eje: hiperplasia suprarrenal congénita (HSC), síndrome de McCune Albrigh, tumores secretores de estrógenos.
- •
Causas genéticas: mutaciones en la proteína GPR 54.
Pubertad precoz periférica: corresponde a aquellas formas de pubertad precoz que no se han originado por activación del eje hipotálamo-hipófisis y, por tanto, son GnRH independientes. Sus causas más frecuentes son:
- •
Tumores ováricos productores de estrógenos: tumores de la granulosa, de la teca, disgerminomas, teratomas. Representan el 11% de las pubertades precoces de origen periférico.
- •
Quistes funcionales.
- •
Alteraciones suprarrenales: tumores productores de estrógenos, HSC.
- •
Síndrome de McCune Albrigh.
- •
Aporte exógeno de estrógenos: alimentos, fármacos, cremas.
- •
Hipotiroidismo primario.
Evolución de la pubertad precoz: a veces, el patrón de acontecimientos es similar a la secuencia evolutiva normal, pero no es infrecuente que no se atenga estrictamente a la cronología puberal fisiológica.
TratamientoObjetivos
- •
Tratamiento del proceso de base.
- •
Detener la maduración ósea.
- •
Frenar los caracteres sexuales iniciados.
- •
Maximizar la talla.
- •
Reducir los problemas psicológicos y sociales que provoca.
Tratamiento médico:
- •
Acetato de ciproterona: presenta un efecto antigonadotropo y bloquea los receptores androgénicos. Dosis: 50-75mg/m2 de superficie corporal/día. Vía oral. Actualmente, poco utilizado debido a sus efectos secundarios y a la escasa mejoría sobre la talla final.
- •
Análogos de la GnRH: involución de los caracteres sexuales secundarios y de la maduración útero-ovárica. Disminución de la secreción de gonadotropinas y esteroides. Disminución de la velocidad de crecimiento y de la maduración ósea. Mejoría del pronóstico de talla final. Efectos secundarios: los dependientes de la vía de administración y, no está confirmado, una posible desmineralización. Los preparados más empleados son:
- ∘
Intranasal: nafarelina 6 pulverizaciones día (1.200μg).
- ∘
Subcutáneo o intramuscular: leuprolida. Inyección mensual de 7,5mg o trimestral de 22,5mg.
- ∘
Triptorelina inyectable: 3,75mg mensual o 11,25mg trimestral.
- ∘
El control del tratamiento se hace mediante evaluación periódica de LH y esteroides, con una primera determinación aconsejable al 2.°-3.° ciclo si se utilizan fármacos depot.
Hay que hacer un control clínico de la talla y de la edad ósea cada 6 meses.
Se desconoce el momento óptimo de suspensión del tratamiento. Los criterios más aceptados son:
- •
A los 11 años de edad cronológica.
- •
Cuando la edad ósea y cronológica se correspondan.
- •
Pronóstico de talla en el momento de la suspensión.
- •
Impacto personal en la restauración de la pubertad.
El pronóstico está en relación con la causa: en las alteraciones del SNC, dependerá de sus posibilidades terapéuticas, mientras que en los tumores malignos dependerá de sus características y evolución. Los tumores benignos y quistes funcionales presentan generalmente buen pronóstico y en los casos de hipotiroidismo primario se produce la normalización tras el tratamiento.
La pubertad normal suele reiniciarse, incluyendo la menarquia, en los 16 meses posteriores al tratamiento.
Tratamiento quirúrgico: en todos los tumores ováricos, suprarrenales o cerebrales que lo requieran. También estaría indicado el tratamiento quirúrgico en los quistes benignos si persisten o se aceleran los signos puberales.
Formas especiales de pubertad precoz (pubertad precoz incompleta)Telarquia prematura (precoz): desarrollo mamario aislado uni o bilateral en niñas menores de 8 años en ausencia de otros caracteres sexuales secundarios.
- •
Exploración física: la talla corresponde a la edad.
- •
Rx edad ósea: no está acelerada.
- •
Laboratorio:
- ∘
Estrógenos: ligeramente mayor a los correspondientes por edad.
- ∘
FSH-LH: normales.
- ∘
Test de la GnRH: valores de gonadotropinas en rango prepuberal.
- ∘
- •
Seguimiento: control clínico cada 6 meses.
- •
Inicialmente no tratamiento.
- •
Pronóstico: 50% estabilización, 30-35% regresión espontánea, 10-20% progresión.
Adrenarquia prematura: aparición de vello pubiano antes de los 8 años de edad. Ocasionalmente asociado a vello axilar, sudoración y acné
- •
Exploración física: correspondencia de talla y edad cronológica.
- •
Rx edad ósea: no está acelerada.
- •
Analítica: moderado aumento de esteroides suprarrenales
- •
Seguimiento: descartar HSC o tumor suprarrenal.
- •
No tratamiento.
- •
Control clínico: cada 6 meses
Menarquia aislada: sangrado uterino cíclico sin otro signo de maduración. La edad ósea es igual a la cronológica. En la analítica, podemos observar un aumento leve de FSH. No presenta repercusiones posteriores.
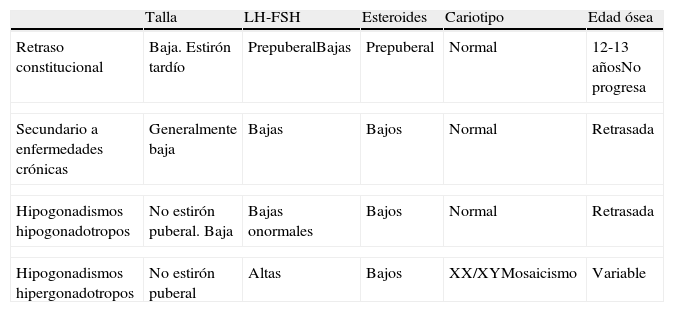
Pubertad retardada (tabla 1)DefiniciónSe considera pubertad retrasada cuando no se ha iniciado el desarrollo de los caracteres sexuales secundarios a los 14 años de edad (equivalente a una edad cronológica superior a 2 desviaciones estándar por encima de la media poblacional). También se incluye en este concepto cuando, tras el inicio del desarrollo de los caracteres sexuales secundarios, pasan 2 años sin la aparición de la menarquia. Se refiere a niñas que a los 14 años no han iniciado el desarrollo de caracteres sexuales o que tras 4-5 años del inicio de los caracteres sexuales no han tenido la menarquia.
Características clínicas y analíticas de la pubertad retrasada
| Talla | LH-FSH | Esteroides | Cariotipo | Edad ósea | |
| Retraso constitucional | Baja. Estirón tardío | PrepuberalBajas | Prepuberal | Normal | 12-13 añosNo progresa |
| Secundario a enfermedades crónicas | Generalmente baja | Bajas | Bajos | Normal | Retrasada |
| Hipogonadismos hipogonadotropos | No estirón puberal. Baja | Bajas onormales | Bajos | Normal | Retrasada |
| Hipogonadismos hipergonadotropos | No estirón puberal | Altas | Bajos | XX/XYMosaicismo | Variable |
- •
Retraso constitucional o idiopático: muy raro en niñas. Puede ser esporádico o de aparición familiar.
- •
Retraso secundario a enfermedades crónicas:
- ∘
Endocrinopatías, nefropatías, enteropatías, neuropatías.
- ∘
Enfermedades hematológicas.
- ∘
Enfermedades infecciosas crónicas.
- ∘
Trastornos de la conducta alimentaria.
- ∘
Enfermedades autoinmunes e inmunodeficiencias.
- ∘
Neoplasias.
- ∘
Estrés y ejercicio físico intenso.
- ∘
- •
Retraso por fallo hipotálamo-hipofisario. Hipogonadismos hipogonadotropos
- •
Congénitos:
- ∘
Déficit de GnRH con anosmia (síndrome de Kallman) o sin anosmia.
- ∘
Déficit de gonadotropinas por alteración del receptor de GnRH.
- ∘
Déficit aislado de FSH o LH.
- ∘
Panhipopituitarismo congénito.
- ∘
Malformaciones cerebrales.
- ∘
Asociado a cromosomopatías.
- ∘
Mutaciones genéticas: gen receptor de FGF1 y CHD7.
- ∘
- •
Adquiridos:
- ∘
Tumores cerebrales.
- ∘
Alteraciones infecciosas.
- ∘
Traumatismos y accidentes cerebrovasculares.
- ∘
Iatrogénicos: cirugía-radioterapia.
- ∘
Hipofisitis autoinmune.
- ∘
- •
Retraso puberal por fallo gonadal. Hipogonadismo hipergonadotropo
- •
Congénitos:
- ∘
Alteración de los receptores de gonadotrofinas.
- ∘
Alteración en la síntesis o acción periférica de estrógenos o andrógenos.
- ∘
Síndrome de Turner y mosaicismos.
- ∘
Disgenesias gonadales.
- ∘
- •
Adquiridos:
- ∘
Ooforitis autoinmune.
- ∘
Radio-quimioterapia.
- ∘
Hemocromatosis.
- ∘
Galactosemia.
- ∘
- •
Asociados a síndromes polimalformativos:
- ∘
Síndromes de Noonan, Fanconi, Prader-Willi, Lynch y Laurence-Moon-Bardet.
- ∘
Distrofia miotónica de Steiner.
- ∘
- •
Anamnesis familiar: menarquia materna, infertilidad, enfermedades genéticas.
- •
Anamnesis personal: enfermedades, capacidad olfatoria, régimen alimenticio, desarrollo pondo-estatural, intensidad de actividades deportivas.
- •
Exploración física: peso/talla, estadio puberal, exploración genital, presencia o no de signos corporales de enfermedad orgánica.
- •
Analítica: hemograma, pruebas hepáticas y renales, prolactina, TSH y T4 libre, LH, FSH, estradiol, test de GnRH, IGF1 (marcador de GH).
- •
Cariotipo.
- •
Rx de edad ósea.
- •
Ecografía ginecológica.
- •
Resonancia magnética o TAC craneal.
Objetivos:
- •
Eliminación de la causa responsable.
- •
Corrección de las alteraciones hormonales.
- •
Conseguir el desarrollo de los caracteres sexuales secundarios.
- •
Inducir la maduración de los órganos genitales internos.
- •
Proteger la masa ósea.
- •
Prevenir las alteraciones cardiovasculares.
Tratamiento médico:
- •
De la enfermedad de base.
- •
Retraso constitucional: en este caso es preferible la conducta expectante. En caso de que provoque problemas psicosociales, se puede pautar un tratamiento con estrogenoterapia. El inicio del tratamiento médico será a los 11-12 años de edad ósea o 13-14 de edad cronológica y la duración hasta tener una edad ósea de 13 años o un estadio iii de Tanner.
- •
Hipogonadismos: estrogenoterapia.
Preparados de estrógenos:
- •
Estrógenos equinos conjugados: 0,3mg/día. Vía oral.
- •
Estradiol: 0,25mg/día. Vía oral. Aumento progresivo hasta los 2mg/día.
- •
17β-estradiol: 0,2mg/día. Aumento hasta 2mg/día.
- •
Estradiol transdérmico: 14-15μg/día (1/2 parche de 25μg cada 3 días). Aumento progresivo hasta 50-100μg.
La estrogenoterapia:
- •
Se inicia generalmente a los 12-13 años de edad ósea.
- •
Se incrementa progresivamente la dosis cada 6-12 meses.
- •
Seguimiento cada 6 meses: valoración de talla, edad ósea y estadio puberal.
- •
Se administra sola durante un año. Si se produce sangrado en este periodo, se puede introducir una progesterona; si no, se incorporará de forma cíclica, del 15 al 25.° día del ciclo cuando las dosis estrogénicas sean las correspondientes a un adulto.
Tratamiento quirúrgico: en caso de tumores o de anomalías genitales.
Los Protocolos Asistenciales de la Sociedad Española de Ginecología y Obstetricia pretenden contribuir al buen quehacer profesional de todos los ginecólogos, especialmente los más alejados de los grandes hospitales y clínicas universitarias. Presentan métodos y técnicas de atención clínica aceptadas y utilizadas por especialistas en cada tema. Estos protocolos no deben interpretarse de forma rígida ni excluyente, sino que deben servir de guía para la atención individualizada a las pacientes. No agotan todas las posibilidades ni pretenden sustituir a los protocolos ya existentes en Departamentos y Servicios Hospitalarios.