





La aplasia cutis congénita es una alteración presente al nacimiento, caracterizada por un defecto focal de piel y en ocasiones de estructuras subyacentes, de localización variable pero más frecuente en el cuero cabelludo. La etiología es desconocida y presenta cierto predominio por el sexo femenino. Se presenta como un defecto único o múltiple, de forma aislada o formando parte de síndromes más complejos de carácter familiar y, en menor medida, de carácter espontáneo. El pronóstico y el tratamiento dependen de la extensión y profundidad de las lesiones.
Aplasia cutis congenita (ACC) is a skin defect present at birth affecting a focal area and sometimes underlying structures. Localization varies but the most frequently affected area is the scalp. The etiology is unknown and females are more commonly affected than males. This entity can manifest as a solitary defect or as multiple lesions. ACC can occur as an isolated defect, but can also be associated with other, more complex familial syndromes; sporadic cases are less frequent. Prognosis and treatment depend on the size and depth of the lesions.
La aplasia cutis congénita (ACC) consiste en un defecto focal de piel presente al nacimiento que puede afectar a cualquier parte de la superficie corporal y es más frecuente en el cuero cabelludo.


En la primera descripción de este desorden del desarrollo, realizada por Cordon1 en 1767, el defecto se localizaba en las extremidades inferiores. En 1826 Campbell2 describió por primera vez esta afección localizada en el cuero cabelludo3.
La incidencia de esta enfermedad oscila en 1-3/10.000 nacidos vivos3,4. Todavía no se ha descrito una etiología única, aunque en algunos casos se encuentra asociado a otras malformaciones. La mayoría de los casos son esporádicos, aunque existe influencia familiar, sobre todo de herencia autosómica dominante, y es más infrecuente la herencia autosómica recesiva.
El tratamiento será de inicio conservador, reservando el tratamiento quirúrgico para los casos que cursen con complicaciones o en los que el defecto comprometa una amplia zona o estructuras vitales subyacentes.
Presentamos el caso de un recién nacido con ACC en la calota, sin otras malformaciones asociadas ni historia familiar de lesiones cutáneas.
CASO CLÍNICOGestante de 29 años, que ingresó en nuestro servicio por amniorrexis a las 38 semanas de gestación. La paciente no refería antecedentes médicos ni quirúrgicos de interés, únicamente tabaquismo (2 cigarrillos/día en el momento del ingreso). No existía historia familiar de lesiones cutáneas o malformaciones. Como antecedente obstétrico presentó un aborto espontáneo completo en 2004, que no precisó legrado. El control gestacional fue realizado en el tocólogo del área de salud extrahospitalaria, donde las ecografías obstétricas seriadas fueron normales, los controles analíticos (bioquímica, hemograma y serologías) también normales, con triple cribado y O'Sullivan negativos. El cultivo vaginorrectal no detectó crecimiento para Streptococcus agalactiae. Durante el embarazo no había ingerido fármacos, salvo hierro, ácido fólico y calcio.
Se trasladó a la paciente a dilatación con el diagnóstico de parto en curso, donde los períodos de dilatación y de expulsivo evolucionaron favorablemente. A la exploración se objetivó un defecto de calota fetal, por lo que no se consideró la monitorización interna para el control fetal. El patrón del registro cardiotocográfico fue tranquilizador en todo momento. Se decidió el traslado de la paciente a quirófano para un parto eutócico, por el posible defecto de calota fetal y un correcto control por parte del servicio de pediatría. Nació un varón, con un peso de 2.570g, una puntuación en la prueba de Apgar de 9–10 y con pH de arteria umbilical de 7,30. La placenta era de aspecto normal.
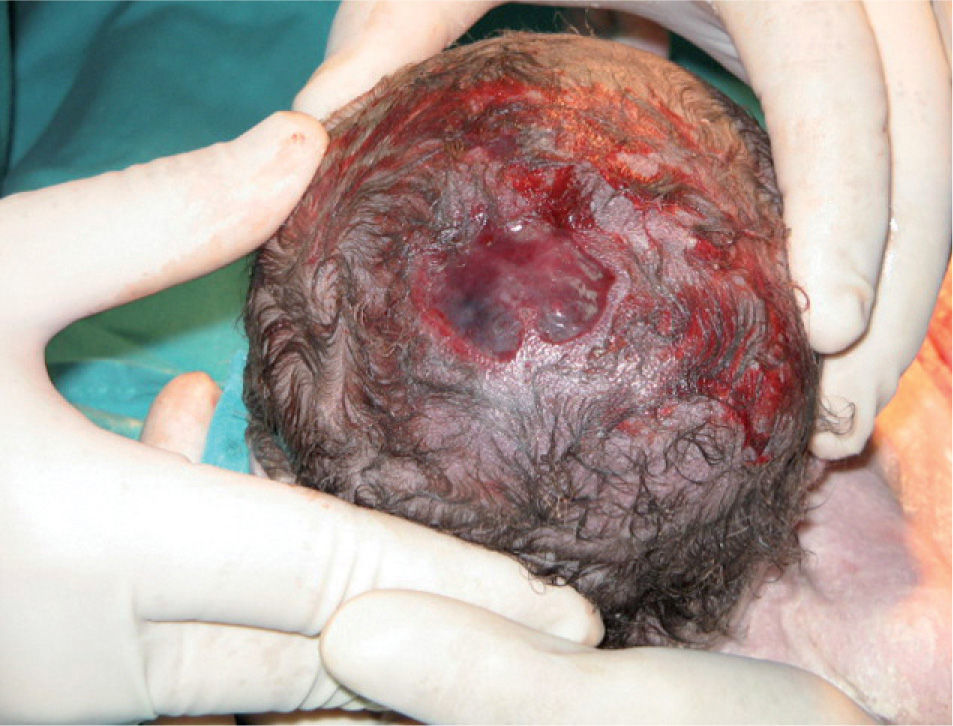
En la exploración inmediata del recién nacido, se observó una aplasia cutis de cuero cabelludo y se decidió su ingreso en el servicio de neonatología. La lesión consistía en una úlcera con bordes en sacabocados, localizada en la región medial de parietales y vértex, de 3cm de diámetro de tamaño, con una zona de 1cm de necrosis superficial en lado izquierdo. El defecto afectaba a la piel y las estructuras subyacentes, con integridad de la duramadre. La piel circundante tenía buen color, sin zonas cruentas (fig. 1). No se observó ninguna otra lesión cutánea y el resto de la exploración física era normal. Se tomaron cultivos del recién nacido (hemocultivo, exudado ótico, frotis nasal, exudado umbilical, exudado ocular) y todos fueron negativos; se realizaron analíticas seriadas, que fueron normales.
Se inició tratamiento conservador, que consistió en limpieza de la zona afectada con solución acuosa de clorhexidina y aplicación posterior de pomada antibiótica (mupirocina) y apósito de Tulgrasum antibiótico para evitar la sobreinfección. Dada la correcta evolución con el tratamiento conservador, se indicó continuar con esta pauta de forma ambulatoria con controles seriados. En posteriores revisiones se observó una buena cicatrización del defecto de calota, que completó la epitelialización a los 2 meses del nacimiento, con alopecia residual en zona occipitoparietal.
Durante su ingreso, se detectó un soplo I-II/IV; mediante ecocardiografía se diagnosticó de comunicación interauricular del ostium secundum de 4mm. En la arteria pulmonar presentaba una ligera aceleración del flujo, no significativa. En controles posteriores el problema cardiológico permaneció estable y se decidió control en 3 años.
DISCUSIÓNLa ACC es una alteración presente al nacimiento, de escasa incidencia y cierto predominio por el sexo femenino5. Aparece como una lesión de tamaño variable caracterizada por la agenesia de epidermis y anejos, con ausencia total o parcial de la dermis. En ocasiones puede afectar a las estructuras subyacentes, como tejido celular subcutáneo, fascia, músculo y hueso, que son hipoplásicos o pueden estar ausentes (fig. 2). Histológicamente se observa una delgada capa de colágeno, sin epitelio suprayacente ni estructuras de anejos cutáneos.
La lesión puede distribuirse por toda la superficie corporal, habitualmente siguiendo la línea media; el 80% se localiza en el cráneo y el vértex es el lugar de aparición más frecuente3,5–7, donde se asocia en un 15-30% de los casos a un defecto óseo craneal e incluso de duramadre. Otras localizaciones son, por orden de frecuencia, brazos, rodillas, ambas caras del tronco y nuca. En un 25% de los casos se asocia la lesión del cuero cabelludo con lesiones en el tronco o las extremidades5,8.
La ACC se presenta como un defecto único o múltiple, de forma aislada o formando parte de síndromes más complejos de carácter familiar y, en menor medida, de carácter espontáneo.
Por ello, toda lesión filiada como ACC debe atenderse desde una doble vertiente: como defecto local susceptible de complicaciones hemorrágicas e infecciosas principalmente4 y, por otro lado, como una manifestación clínica en el contexto de un trastorno que puede asociar alteraciones metabólicas, genéticas o malformaciones estructurales a otros niveles.
Se plantean varias hipótesis que pretenden explicar el origen de este defecto congénito; si bien la etiopatogenia supone un aspecto todavía sin aclarar. Entre las posibles causas se contemplan la presencia de bridas intracavitarias de origen infeccioso (varicela y herpes simple)3, responsables de la creación de adherencias amnióticas con la cabeza fetal y el consecuente arrancamiento cutáneo de esa zona. Sin embargo, la presencia de placentas normales en la mayoría de los casos desacredita esta teoría.
La mayoría de los casos de ACC se describe como lesiones de aparición espontánea, sin otras alteraciones asociadas, si bien cuando el defecto es de localización craneal se ha observado una mayor asociación a un patrón familiar (hasta un 40% de los casos) de herencia autosómica dominante con expresión variable en la mayoría de ellos. Se han notificado otros patrones de herencia. Cuando la ubicación es extracraneal, el carácter hereditario no está tan claro3.
Se cuestiona un origen teratógeno asociado a fármacos como los antitiroideos (metimazol y carbimazol), misoprostol (prostaglandina E2), ácido valproico, heparinas no fraccionadas y benzodiazepinas administradas en primer trimestre3,5,9. Sin embargo, no se encuentra este antecedente en todos los casos.
Stephan et al10 explican la ACC del vértex como el resultado de una rotura de la piel y los tejidos subyacentes por sobredistensión secundaria al rápido crecimiento del cráneo y el cerebro fetal.
Distintos autores han planteado la posibilidad de considerar la ACC como un defecto de cierre del tubo neural. Sin embargo, recientes publicaciones incluyen en este grupo únicamente a una variante del defecto, la aplasia congénita bullosa cuyo contexto clínico y pronóstico es igual al de la ACC, pero con una histología y marcadores bioquímicos similares a los presentes en encefaloceles y mielomeningoceles11,12.
Actualmente, el mecanismo etiopatogénico más aceptado es el de carácter vascular. La ACC se considera una lesión cicatricial de origen isquémico. La alteración vascular puede ser consecuencia de factores genéticos o espontáneos, intrínsecos o extrínsecos. Esta teoría explicaría claramente asociaciones descritas, tales como embarazo gemelar con un feto papiráceo y el otro afecto de ACC (por paso de tromboplastina fetal de una circulación a otra)3, mayor incidencia del defecto en hijos de madres consumidoras habituales de cocaína y anfetaminas (sustancias vasoconstrictoras)3,5.
La imagen de las lesiones de la ACC es muy diversa. Habitualmente, presentan un contorno oval irregular y un tamaño variable que condicionará en gran medida el pronóstico del cuadro. Si el defecto se produce en las primeras fases de la gestación, tras el nacimiento ofrece el aspecto de una zona cicatricial atrófica con una alopecia asociada. Cuando el origen es posterior, se presenta como una úlcera denudada con tejido de granulación en los bordes y una base que depende de la persistencia de los distintos tejidos3. En nuestro caso, la lesión aparece como una úlcera con bordes en sacabocados cuyo fondo lo constituían las meninges, que quedaban al descubierto por la agenesia completa de la región medial de ambos parietales, lo cual ocurre en el 20% de los casos descritos3,5.
La mayoría de los cuadros de ACC se manifiestan como lesiones en la superficie corporal, sin ningún otro desorden asociado. Un 10% de los casos aparece asociado a otras alteraciones. Las más frecuentes son las malformaciones de las extremidades (sindactilia, pie zambo, acortamiento, deformación o agenesia de huesos largos, etc.) y el labio leporino. En menor número se han documentado casos asociados a alteraciones craneales y cerebrales (acrania parcial, hidrocefalia, holoprosencefalia), onfalocele, malformaciones cardíacas (fundamentalmente coartación de aorta13), renales y otras manifestaciones dérmicas (epidermólisis bullosa, alteraciones ungueales, etc.). En un pequeño porcentaje, la ACC asociada a otras manifestaciones se ajusta a la descripción de un síndrome conocido, habitualmente de carácter hereditario, por ejemplo, síndrome de Adams-Oliver (ACC en vértex asociado a defectos transversales de las extremidades), síndrome de Johanson-Blizzard y el síndrome de Bart3,4. El 25% de los pacientes afectados de trisomía 13 y 15 presentan lesiones de ACC5. Sin embargo, la mayoría de estas asociaciones de síntomas no se ajustan a ningún cuadro previamente documentado.
Las dos clasificaciones de la ACC más ampliamente difundidas categorizan el defecto según aparezca de forma aislada o asociado a otras manifestaciones. La clasificación de Sybert14, y posteriormente la de Frieden15, fueron publicadas en 1985 y 1986, respectivamente (tablas 1 y 2). En nuestro caso, la ACC se asocia a una comunicación interauricular, que no aparece en ninguna de ellas por lo que se incluye dentro del tipo I de Sybert o del grupo 1 de Frieden.
Clasificación de Sybert de la aplasia cutis congénita (ACC)
| Tipo | Definición | Herencia |
| I | ACC limitada a cuero cabelludo | Esporádica y familiar (autosómica dominante con penetrancia incompleta, expresión variable intrafamiliar) |
| II | ACC que afecta al cuerpo con o sin afección del cuero cabelludo | Esporádica y familiar (autosómica dominante con penetrancia reducida o autosómica recesiva) |
| IIA | ACC que afecta al cuerpo con o sin afección de extremidades | |
| III | ACC limitada al cuero cabelludo con o sin afección de las extremidades | Esporádica y familiar (autosómica dominante) |
| IV | ACC asociada a epidermólisis bullosa (letal, distrófica o simple) | |
| IVA | Síndrome de Bart, con defectos cutáneos localizados, ampollas recidivantes en piel y mucosas y alteraciones ungueales | Autosómica dominante |
Clasificación de Frieden de la aplasia cutis congénita (ACC)
| Grupo | Definición | Herencia |
| 1 | ACC en cuero cabelludo sin anomalías múltiples | Autosómica dominante y esporádica |
| 2 | ACC en cuero cabelludo con anomalías de las extremidades (extremidades reducidas, sindactilia, pie zambo, falta o distrofia de uñas) | Autosómica dominante |
| 3 | ACC en cuero cabelludo con nevos epidérmicos u organoides asociados | Esporádica |
| 4 | ACC sobre malformaciones embrionarias (mielomeningocele, disrafia espinal, gastrosquisis, angiomatosis leptomeníngea, estenosis craneal) | Depende de la afección fundamental |
| 5 | ACC con feto papiráceo o infartos placentarios asociados | Esporádica |
| 6 | ACC asociada con epidermólisis ampollar | Depende del tipo de epidermólisis ampollar |
| 7 | ACC localizada en extremidades sin aparición de vesículas | Autosómica dominante o recesiva |
| 8 | ACC causada por teratógenos (metimazol, varicela, herpes simple) | No es hereditaria |
| 9 | ACC relacionada con síndromes de malformación (trisomía 13, síndrome 4p, síndrome de Johanson-Blizzard, displasias ectodérmicas, hipoplasia dérmica focal, síndrome de bandas amnióticas) | Depende del síndrome |
El diagnóstico de la ACC suele ser posnatal, ya que su identificación mediante ecografía prenatal resulta muy complicada; no obstante, debe incluirse en el diagnóstico diferencial de gestaciones con alfafetoproteína y seudocolinesterasa aumentadas en suero materno y líquido amniótico con valores de acetilcolinesterasa y ecografía normales11.
Tanto el pronóstico como la conducta terapéutica estarán condicionados por el tamaño, la profundidad y la ubicación de la lesión, así como por la presencia de malformaciones asociadas.
Las lesiones de pequeño tamaño que no dejan al descubierto estructuras comprometidas y tienen baja probabilidad de complicaciones se tratan de forma conservadora con curas locales oclusivas y antibioterapia tópica o sistémica. La reepitelialización a partir de los bordes del defecto tarda en completarse de 1 a 3 meses después del nacimiento, con alopecia residual posterior. Se debe valorar la posibilidad de cubrir el defecto con colgajos locales con el fin de acelerar la recuperación y mejorar el resultado estético. El tratamiento de las lesiones amplias con defectos óseos asociados o con alto potencial de complicaciones graves es, sin embargo, un punto controvertido hasta el momento, debido a la escasa experiencia con los resultados. La conducta conservadora conlleva una mortalidad del 20%3 secundaria a hemorragias y tromboembolias sépticas del seno venoso sagital. Por otra parte, el tratamiento quirúrgico agresivo y temprano mediante craneoplastias e implantes cutáneos implica un riesgo importante de hemorragia cerebral intra y postoperatoria inmediata16.