







El complejo Dandy-Walker es una malformación congénita esporádica originada por una disembriogénesis del cerebro medio, que conlleva un grado variable de alteraciones anatómicas en el cerebelo y el cuarto ventrículo principalmente. Puede estar asociado o no a otras malformaciones.
La presencia de otras anomalías se asocia a un aumento significativo de la morbilidad neurológica y la mortalidad neonatal.
Presentamos el caso de una gestante marroquí que no acude al control ecográfico de la semana 20, en la ecografía del tercer trimestre se objetiva un retraso de crecimiento intrauterino precoz, síndrome Dandy-Walker y malformaciones en las extremidades.
Dandy-Walker complex is a sporadic congenital malformation of the midbrain dysmorphogenesis that involves a variable degree of anatomical abnormalities mainly in the cerebellum and fourth ventricle. This syndrome can be associated with other malformations.
The presence of other anomalies is associated with significantly higher neurological morbidity and neonatal mortality.
We report the case of a pregnant Moroccan woman who did not attend the 20th week ultrasound prenatal visit. The third trimester ultrasound scan showed intrauterine growth retardation, Dandy-Walker syndrome, and deformities of the extremities.
La malformación de Dandy-Walker clásica se caracteriza por el agrandamiento de la cisterna magna, defectos del vermis cerebeloso, a través del cual, el quiste se comunica con el iv ventrículo, también aumentado1. Es un desarrollo anormal del sistema nervioso central que puede estar asociado a otras alteraciones cerebrales y extracraneales.
La incidencia de Dandy-Walker es de 1/25.000-35.000 nacidos vivos2 y es la causa del 10% de los casos de hidrocefalia. La tasa de mortalidad por esta alteración alcanza un 12-50% y, asociada con otras malformaciones congénitas, constituye el 83% de la mortalidad posnatal. Aproximadamente, el 70-90% de los pacientes presentan hidrocefalia, que suele desarrollarse tras el nacimiento.
Caso clínicoPresentamos el caso de una secundigesta de nacionalidad marroquí, de 24 años de edad, sin antecedentes familiares ni personales de interés, que tuvo un parto eutócico en 2004, sin incidencias. Grupo y Rh B+, con serologías negativas a toxoplasma, virus de la hepatitis C, Ag HBs, virus de la inmunodeficiencia humana y VDRL y inmunización frente a rubéola.
Primera ecografía, realizada a las 11 semanas de gestación, acorde y normal. La paciente no acude a la ecografía del segundo trimestre.
Ecografía a las 31 + 3 semanas: acorde con 25 + 4 semanas de gestación, se objetiva una cisterna magna de 11mm, con separación del vérmix cerebeloso, microcefalia (fig. 1) y malformación de los dedos de la mano, con falanges incurvadas y dedos rígidos (camptodactilia) (fig. 2); se ofrece la posibilidad de realización de amniocentesis, que acepta, y se cita para resultados en 2 semanas.


Resultado de amniocentesis: 46 XY, cariotipo normal.
Ecografía a las 32 + 5 semanas: acorde con 27 + 2 semanas, PFE: 1.097 g, pelvis renales de 8 y 13mm, fosa posterior de 11,5mm. Se realizan Doppler AU y ACM, que resultan ser normales. Se ofrece la posibilidad de interrupción de gestación por sospecha de síndrome polimalformativo, pero los padres deciden continuar con el embarazo.
Ecografía a las 34 + 4 semanas: acorde con 28 + 2 semanas, PFE: 1.245 g. Manos malformadas y pies toscos«en mecedora» (fig. 3). Malformación de Dandy-Walker con cisterna magna de 22mm, sin cambios. Doppler AU y ACM, normales.

Ecografía a las 37 semanas: acorde a 29 + 2 semanas de gestación, PFE: 1.413 g. Doppler AU y ACM normal, crecimiento intrauterino retardado precoz, Dandy-Walker, iv ventrículo de 18mm, cisterna magna de 24mm, microcefalia, ambas manos malformadas, con falanges acortadas y carpo deforme, aspecto simiesco. Se remite a paritorio para RCTG.
En el paritorio se realiza RCTG durante 40 min, donde se objetiva ausencia total de variabilidad, tanto a corto como a largo plazo, sin aceleraciones ni desaceleraciones y sin dinámica uterina. Se pone en conocimiento del servicio de Pediatría, quienes junto con el servicio de Ginecología y Obstetricia informan a los padres de la posible muerte fetal intraparto o en el posparto inmediato por el síndrome polimalformativo que presenta el feto. Se decide inducción del parto por ser la mejor opción materna y el mal pronóstico fetal.
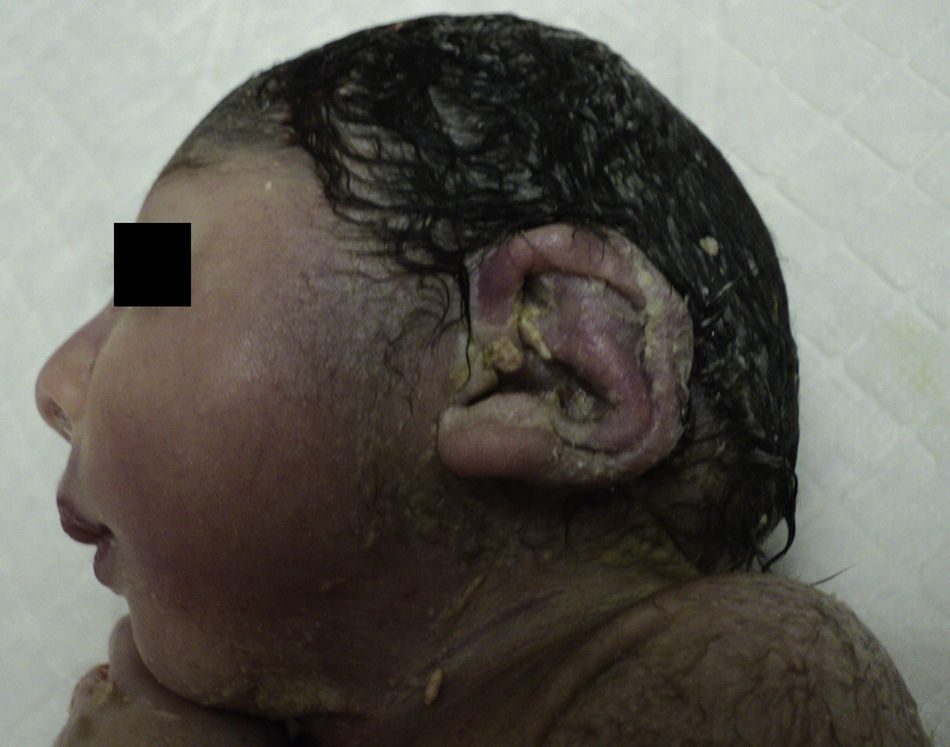
Nace un varón, de 1.370 g, Apgar 1,0,0, pH 7,33, con varias malformaciones externas: microcefalia, orejas de implantación baja y malformadas (fig. 4), raíz nasal ancha, retrognatia, manos malformadas y pies en mecedora (fig. 5), criptorquidia, hipertonía generalizada (fig. 6), con hipoplasia de extremidades inferiores compatible con síndrome polimalformativo.



Se ofrece a los padres la realización de necropsia, pero por sus creencias religiosas es rechazada.
DiscusiónLa malformación de Dandy-Walker es un desorden congénito que se caracteriza por la disgenesia cerebelar, dilatación quística del cuarto ventrículo y elongación de la fosa posterior, con desplazamiento superior de los senos durales y el tentorio cerebeloso. Dicha anomalía es más frecuente en el sexo femenino, con una relación 3:1, su incidencia es de 1/25.000-30.000 nacimientos y engloba un 10% de los casos de hidrocefalia.
El término de complejo de Dandy-Walker se emplea para designar anomalías de la fosa posterior:
- –
Malformación clásica de Dandy-Walker: caracterizada por una cisterna magna aumentada y la agenesia completa o parcial del vermis cerebeloso y un tentorio elevado.
- –
Variante Dandy-Walker: hipoplasia de grado variable de grado variable del vermis, con o sin aumento de la cisterna magna.
- –
Megacisterna magna: integridad del vermis y del cuarto ventrículo.
Fue descrito por primera vez en 1887 por Sultton3, que describió la hipoplasia cerebelar y el quiste en la fosa posterior; en 1914 Dandy y Blackfan4 describieron la tríada típica del síndrome. Y en 1942, Taggart y Walker5 lo relacionaron con la presencia de atresia de los agujeros de Luschka y Magendie. En 1984, Hirsch et al.6 realizaron una revisión de 40 pacientes, que mediante estudios radiológicos, describieron las variaciones anatómicas existentes, destacando también malformaciones en otros órganos y sistemas que podrían estar presentes.
Aproximadamente, el 68% de los enfermos presenta una o más malformaciones asociadas7, que pueden estar situadas dentro o fuera del sistema nervioso central y que tienen una gran repercusión negativa sobre la evolución de los pacientes. Entre las anomalías neurales más frecuentes se encuentran: microcefalia, agenesia del cuerpo calloso, trastornos de migración de los cerebelos, estenosis del conducto de Silvio, espina bífida, siringomielia8. Las no neurológicas incluyen anomalías craneofaciales, displasias óseas, alteraciones cardiacas y urogenitales. Entre los defectos en las extremidades asociados a este síndrome se incluyen sindactilia, clinodactilia, camptodactilia y acortamiento de falanges2.
La anomalía más frecuente extracraneal son los defectos cardiacos, que están presentes en un 29% de los casos, y que incluyen defectos en el septo ventricular, persistencia del ductus arterioso, trasposición de grandes vasos y estenosis pulmonar congénita. En frecuencia menor, se encuentran los defectos en las extremidades y renales como el reflujo vesicoureteral, el hidrocele y el riñón en herradura9.
Se describen síndromes complejos polimalformativos: son los síndromes de Aicardi, de Ellis-Van Creveld, de Fraser, de Fryns, de Marden-Walken, de Meckel, de Neu-Laxova, oro-facio-digital y de Walker-Walburg5,10-12; todos tienen en común la malformación de Dandy-Walker. También está asociado a anomalías cromosómicas, incluyendo las trisomías 13 y 18, y el síndrome de Turner. Se ha relacionado con ciertos teratógenos, alcohol e infecciones virales, aunque no existe una evidencia clara.
La ecografía prenatal es un método básico para el diagnóstico y para la evaluación y la asociación con otras anomalías1. Si el diagnóstico prenatal no es posible, los síntomas normalmente aparecen en la temprana infancia e incluyen un lento desarrollo psicomotor, abombamiento de la fontanela anterior, hidrocefalia y progresivo alargamiento del cráneo. Según progresa el desarrollo, los síntomas más severos son los asociados al aumento de la presión intracraneal, como son la irritabilidad, los vómitos y los signos de disfunción cerebelar, las alteraciones de la marcha y la falta de coordinación muscular. Los efectos de dicha malformación en el desarrollo intelectual son muy variables; la mitad tiene un desarrollo cognitivo normal y su asociación con otras malformaciones empeora el pronóstico de los pacientes afectados.
El diagnóstico en la edad adulta es poco frecuente y suele ser un hallazgo casual. En estadios avanzados puede causar la muerte la compresión del tronco del encéfalo o por herniación a través de los agujeros de Luschka y Magendie.
Una alta proporción de las muertes neonatales tienen en común la afectación de varios órganos, además de la malformación de Dandy-Walker. Niños con dicha anomalía que presentan defectos al nacimiento en los que están afectados 2 o más órganos tienen peor tasa de supervivencia, como ha ocurrido en el caso que presentamos.
Se han realizado estudios en los que la mortalidad posnatal es 6 veces superior en niños con Dandy-Walker que presentan 2 o más anomalías en otros órganos comparados con aquellos que presentan dicha malformación de forma aislada13,14.
Ante la presencia de un síndrome polimalformativo asociado a Dandy-Walker, sería necesaria la realización de una necropsia para poder identificarlo. La amniocentesis descartó anomalía cromosómica asociada, pero no fue posible clasificar al paciente debido a la negativa de los padres de realización de necropsia por sus creencias religiosas.
Lo más probable es que la causa de muerte en el posparto inmediato del neonato fuera la malformación intracraneal o Dandy-Walker, asociada a otras malformaciones mayores y menores, y al retraso del crecimiento intrauterino, que le ocasionaba una gran inmadurez. Disfunciones en el tronco del encéfalo ocasionan dificultades en la succión que pueden ocasionar vómitos y aspiración. La anormalidad severa del vermis se asocia a otras malformaciones del encéfalo que siempre se acompañan de retraso mental. La muerte prematura usualmente es debida a defectos congénitos cardiacos severos y a distintos grados de hipoplasia pulmonar.
Responsabilidades éticasProtección de personas y animales. Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.