


La lisencefalia es una malformación cerebral caracterizada por la ausencia (agiria) o reducción (paquigiria) de las circunvoluciones cerebrales, causado por una migración neuronal anómala en el neocórtex. Presentamos un caso de sospecha ecográfíca prenatal de feto afectado de lisencefalia, confirmado mediante resonancia magnética posnatal. El recién nacido presentó una dismorfia facial característica y el cariotipo con estudio FISH una deleción del cromosoma 17p13.3, lo que nos llevó a un diagnóstico definitivo de síndrome de Millar-Dieker.
Lissencephaly is a cerebral malformation characterized by the absence (agyria) or reduced number (pachygyria) of convolutions, caused by a failure of neuron migration in the neocortex. We present the case of a fetus with a suspected diagnosis of lissencephaly based on prenatal ultrasound, which was confirmed by postnatal magnetic resonance imaging. The newborn showed characteristic facial dysmorphism, and cytogenetic analysis with FISH studies showed a deletion of a region at chromosome locus 17p 13.3, leading to a definitive diagnosis of Miller-Dieker syndrome.
La lisencefalia es un raro desorden del desarrollo cerebral que se caracteriza por la ausencia (agiria) o reducción (paquigiria) de las circunvoluciones cerebrales, causado por una migración neuronal anómala en el neocórtex1.
Las manifestaciones clínicas de esta anomalía incluyen retraso psicomotor severo, convulsiones y retraso en el desarrollo2.
La lisencefalia puede presentarse de forma aislada o asociada con varios fenotipos; uno de ellos es el síndrome de Miller-Dieker (SMD), desorden genético que consiste en la deleción del cromosoma 17p 13.3. Se caracteriza por presentar el grado más severo de lisencefalia con dismorfismo facial asociado; también pueden estar presente otras malformaciones, como el onfalocele, anomalías cardiacas, genitourinarias, pie equino-varo, etc.3. Su pronóstico es muy malo, limitándose la supervivencia a los 6 primeros años de vida, a lo sumo.
Clásicamente, el diagnóstico prenatal de la lisencefalia se basa en la ausencia de circunvoluciones visibles en la ecografía y, puesto que éstas no se desarrollan hasta finales del segundo trimestre, el diagnóstico será tardío1. Actualmente, con el patrón de desarrollo de las cisuras cerebrales descrito por Toi et al4 es posible realizar un diagnóstico más temprano.
Otros hallazgos ecográficos presentes en el SMD son la ventriculomegalia, el polihidramnios, el retraso del crecimiento y la presencia de otras malformaciones asociadas.
En caso de sospecha ecográfica de lisencefalia deberían realizarse, como exploraciones complementarias, una resonancia magnética (RM) y un estudio genético que incluya un análisis FISH para detectar una posible deleción en el cromosoma 17p 13.32,3,5-7.
En caso de confirmarse el diagnóstico de SMD, se recomienda un manejo obstétrico conservador, dado el mal pronóstico de esta entidad1,6,7.
Caso clínicoSecundigesta de 35 años que nos es remitida a la unidad de ecografía en la semana 34 de gestación ante el hallazgo de un polihidramnios y un estómago visible a escasa repleción. Antecedentes personales y familiares sin interés. Embarazo y parto anterior, normales. La gestación actual había sido controlada y de curso normal. El cribado combinado del primer trimestre fue informado de «bajo riesgo» de síndrome de Down. En la ecografía de la semana 20 se destacó que el estómago tardó en ser visualizado. La medida del atrio ventricular fue normal (DTA=7,7mm). La paciente se citó para nuevo control pero no acudió a la cita programada.
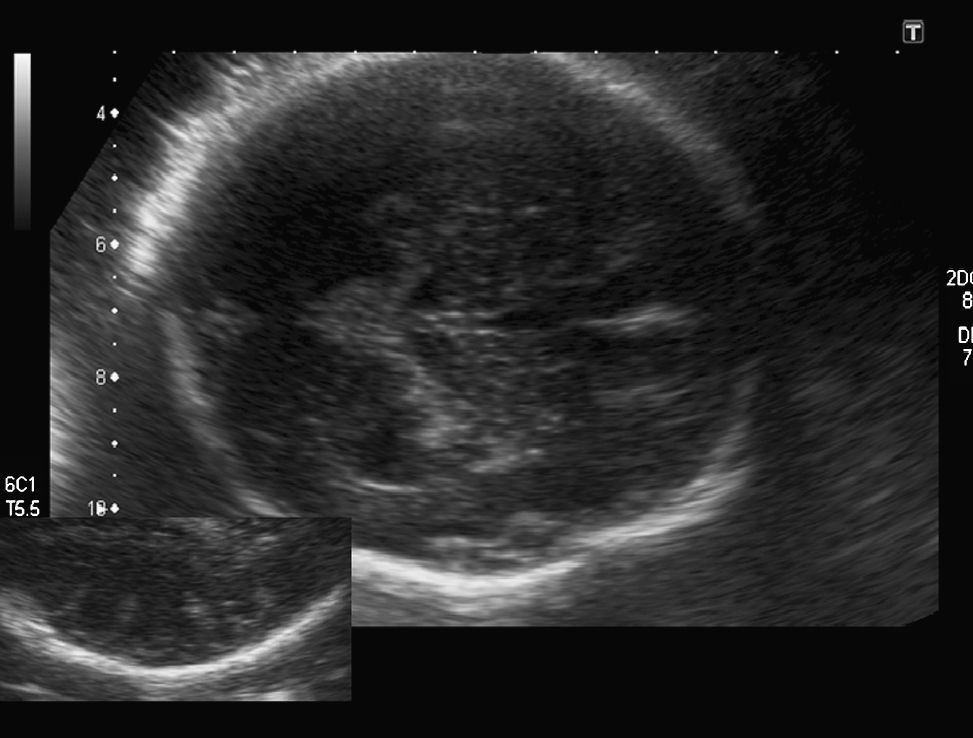
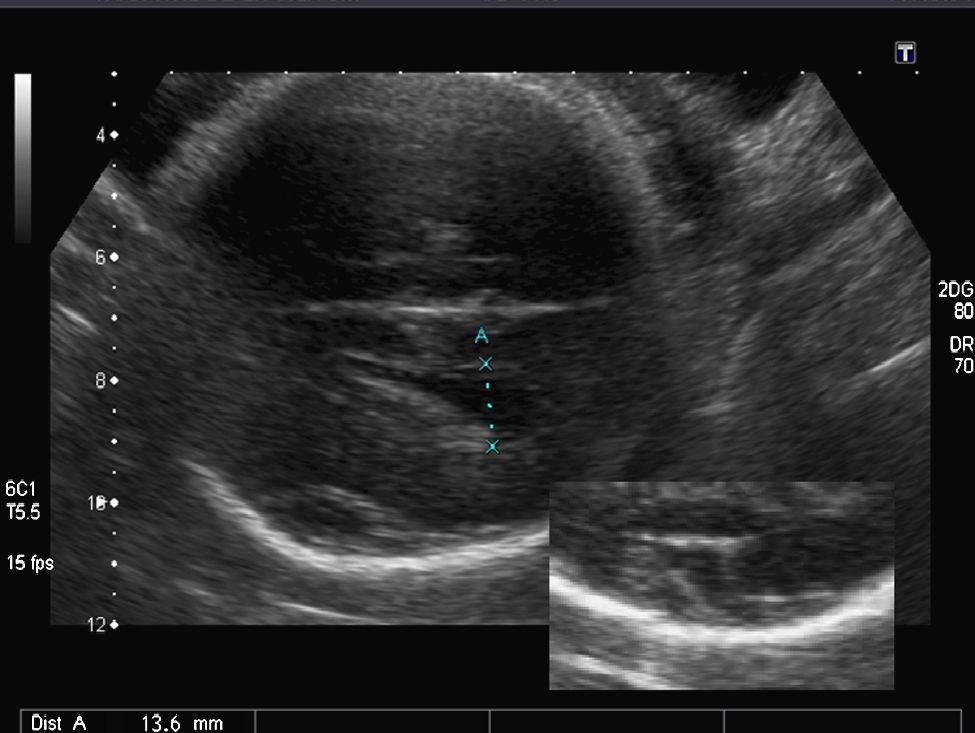
Hallazgos en la ecografía prenatal: ausencia total de cisuras y circunvoluciones, dando un aspecto de «cerebro liso» (fig. 1). La cisura de Silvio presentaba una morfología anómala para la edad gestacional. Ventriculomegalia bilateral moderada (DTA=14,4mm) y discreto agrandamiento del tercer ventrículo (fig. 2). El resto de las estructuras intracraneales se visualizaron normales. El estómago era visible, aunque a escasa repleción. El resto de los órganos fetales fueron vistos normales. El peso fetal estimado estaba en el p. 10-25 y la circunferencia cefálica en el p. 10 para las 34 semanas de gestación. El índice de líquido amniótico fue de 41cm.


El diagnóstico de sospecha fue el de lisencefalia. Los padres fueron informados sobre el mal pronóstico de esta malformación y les recomendamos la realización de una RM para confirmar el diagnóstico y de una amniocentesis genética, pero rechazaron ambas propuestas ante el avanzado estado de la gestación.
El parto eutócico se produjo en la semana 38. La puntuación en el Apgar al nacimiento: 4-9. El peso fue 2.520g (p. 10-25), la talla 47cm (p. 25-50) y el perímetro craneal 32cm (p. 10). La recién nacida presentaba unos rasgos faciales característicos (frente prominente, ojos pequeños, boca «en carpa», implantación baja del pelo), hipotonía y mala coordinación succión-deglución.
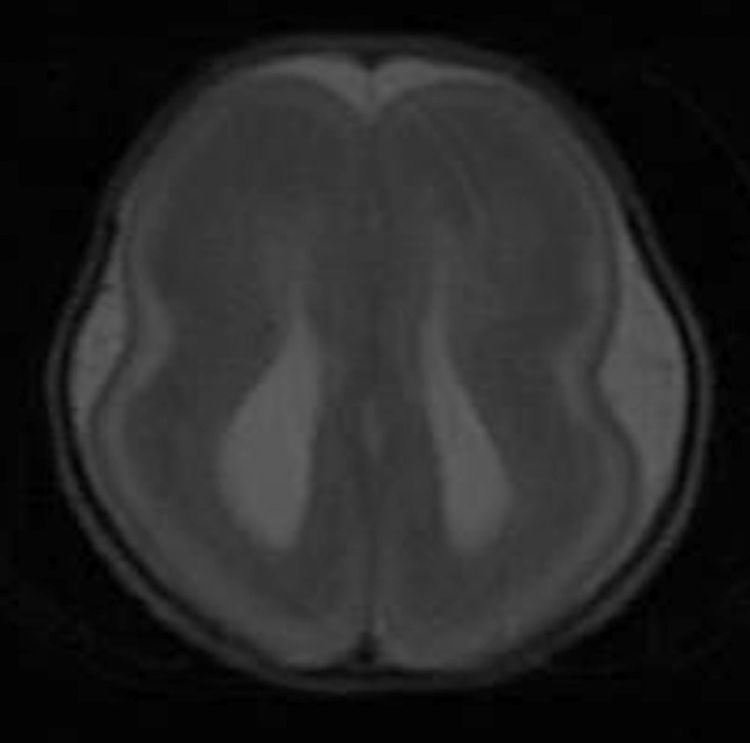
Los hallazgos en la RM posnatal eran compatibles con lisencefalia completa, con ausencia del patrón de surcación normal en el parénquima cerebral (fig. 3).

El cariotipo fue 46 XX 9qh++ (variante polimórfica sin repercusión fenotípica). En el estudio FISH: deleción de la región 17 p 13.3, donde se localiza el gen LIS1 que se asocia al SMD.
Actualmente, la niña tiene un año de vida, presenta un gran retraso psicomotor y del desarrollo, ataques convulsivos frecuentes, a pesar del tratamiento, y ha ingresado en el hospital en varias ocasiones por neumonías de repetición.
DiscusiónEl córtex cerebral se desarrolla en tres etapas que se superponen: la proliferación y diferenciación de los precursores neuronales, la migración neuronal y la formación y organización del córtex en láminas. La migración neuronal es un proceso secuencial que dará lugar a un córtex organizado en 6 capas. La formación de las cisuras y circunvoluciones ocurre al final de la migración y es consecuencia de un rápido crecimiento de la superficie cortical externa sin un crecimiento similar por parte de la interna. La disrupción de estos estadios del normal desarrollo resulta en un amplio espectro de malformaciones del córtex cerebral8.
La lisencefalia («cerebro liso») es una malformación severa del córtex cerebral, resultado de un fallo de la migración neuronal durante el tercer y cuarto mes de gestación. El cerebro afectado muestra la ausencia (agiria) o escasez (paquigiria) de cisuras y circunvoluciones2.
Sobre la base de los hallazgos clínico-patológicos, la lisencefalia se ha dividido clásicamente en dos grupos:
- –
Tipo I o lisencefalia clásica: el córtex normal, constituido por 6 capas, es reemplazado por un córtex anormalmente grueso, remodelado en 4 capas. La lisencefalia clásica se puede presentar como lisencefalia aislada o asociada al SMD o al síndrome de Norman-Roberts.El SMD presenta dismorfismo facial asociado y el grado más severo de lisencefalia, por lo que es más fácil de reconocer prenatalmente, debido a la llamativa falta total de cisuras y circunvoluciones.
- –
Tipo II: se caracteriza por un córtex desorganizado, no estructurado en capas. Se observa en algunos casos de distrofia muscular congénita1,2,6,7.
La aparición progresiva de las cisuras y las circunvoluciones cerebrales en la ecografía pueden permitir estimar el grado de maduración cerebral en el feto2.
Toi et al4 han descrito el patrón ecográfico de desarrollo de las cisuras cerebrales entre la 15,6 y la 29,6 semanas de gestación. Este patrón incluye las cisuras primarias en la superficie media de los hemisferios (fisura parieto-occipital, fisura calcarina y el sulcus cingulado), las cisuras de la superficie lateral convexa de los hemisferios y el proceso de operculización de la ínsula o cisura de Silvio. La fisura parieto-occipital siempre debe ser visible a partir de las 20,5 semanas de gestación; la fisura calcarina a partir de la 21,9 semanas y el sulcus cingulado a partir de las 24,3 semanas. Las cisuras de la superficie lateral convexa de los hemisferios siempre serán visibles a partir de las 27,9 semanas. La ínsula formará un ángulo agudo con el opérculo del lóbulo temporal que deberá ser siempre visible después de las 24,5 semanas de gestación4.
La importancia del conocimiento de este patrón de desarrollo prenatal de la surcación radica en que permite realizar el diagnóstico de algunos desórdenes de la migración de forma temprana, utilizando cortes estandarizados en el cribado ultrasonográfico, sin necesidad de un entrenamiento extenso8.
Fong et al3, en un estudio retrospectivo, encontraron que el diagnóstico de lisencefalia asociada a SMD podría haber sido sospechado antes de la semana 24 de gestación en base a la ausencia de la fisura parieto-occipital y calcarina y a una apariencia anómala de la cisura de Silvio.
En nuestro caso, el diagnóstico fue tardío, en el tercer trimestre avanzado (nuestro protocolo de control de embarazo de bajo riesgo sólo incluye una ecografía por trimestre). La ausencia de visualización de todas las cisuras y circunvoluciones, así como una morfología anómala de la cisura de Silvio, nos hizo sospechar el diagnóstico de lisencefalia. El haber hecho una ecografía más precoz y, siguiendo el patrón descrito por Toi, seguramente hubiese permitido realizar un diagnóstico más temprano pero poco hubiese variado el manejo de la gestación, puesto que la legislación española no permite la interrupción de la gestación más allá de la semana 22. Quizá sí nos habría permitido realizar una RM y un estudio genético prenatal que nos habrían dado el diagnóstico exacto de la entidad.
En el SMD, además de la agiria, hay otras anomalías intracraneales presentes, como la ventriculomegalia moderada, el espacio subaracnoideo prominente y, en algunos casos, la agenesia del cuerpo calloso7. La ventriculomegalia no es un hallazgo específico; sin embargo, es un importante marcador y un hallazgo prenatal común3,6,7. En el estudio de Fong, la ventriculomegalia moderada estuvo presente en 6 de los 7 fetos con SMD en el primer examen ecográfico que fue realizado entre la 20 y la 33 semanas de gestación3. En nuestro caso, la ventriculomegalia estuvo presente en el momento del diagnóstico; sin embargo, la medida del atrio ventricular fue normal en la ecografía de la semana 20.
Otro hallazgo asociado al SMD casi constante en la ecografía prenatal es el polihidramnios. Se cree que es el resultado de una depresión severa del SNC que conlleva dificultades en la deglución o una deglución disminuida6,7. Al igual que en el caso comunicado por Lenzini5, este hallazgo, junto con la visualización de un estómago a escasa repleción, fue la primera observación detectada en nuestro caso.
En general, la actividad fetal está disminuida1,7. En algunos casos se ha documentado una actividad agitada por parte del feto1.
También es común la restricción del crecimiento intrauterino3,6. En algunas series llega a ser del 50%7, aunque no está presente en otros casos comunicados1,6. Algunos autores indican que la restricción del crecimiento es un fenómeno posnatal y que la circunferencia cefálica puede ser normal o algo más pequeña al nacimiento aunque su crecimiento se enlentecerá después1. En nuestro caso, tanto el peso como la circunferencia cefálica fetal estaban en percentiles bajos.
Respecto a las anomalías faciales asociadas con el SMD (hipoplasia mandibular, micrognatia, etc.), la mayoría de las veces son sutiles y pasan desapercibidas en la ecografía prenatal3.
Otras malformaciones asociadas al SMD incluyen: anomalías cardíacas, pie equino-varo, polidactilia, atresia duodenal y onfalocele3,5-7. Ninguna estuvo presente en nuestro caso.
La RM posnatal es el método de elección para el diagnóstico de una anomalía del desarrollo cortical2,9. La RM es superior a los ultrasonidos en la detección de la lisencefalia, proporciona una representación más completa y global de las cisuras cerebrales puesto que la visibilidad no está restringida por los huesos craneales y la posición fetal2. En el SMD las imágenes axiales proporcionan una apariencia del cerebro en «reloj de arena» o «figura en 8». Además, la RM permitirá también la detección o confirmación de otras anomalías asociadas, tanto craneales como extracraneales2,3,9. Aunque en nuestro caso no la realizamos prenatalmente (carecemos de ella en nuestro centro), pensamos que debería realizarse ante cualquier sospecha de malformación del SNC, ya que permite confirmar o no el diagnóstico, así como mejorar o reafirmar la información transmitida a los padres.
El SMD se asocia con una deleción del locus 17p 13.32,3. Entre los genes mapeados en este locus se encuentra el gen LIS1, cuya mutación es la causante de la lisencefalia5. Otros genes adicionales, como el 14-33 épsilon, DSX, RELN y ARX parecen determinar el dismorfismo facial y la forma más severa de lisencefalia5,8. Por tanto, ante la sospecha ecográfica de lisencefalia debería realizarse un estudio cromosómico que incluyese un análisis FISH con la sonda de ADN específica para detectar deleciones en el cromosoma 17p 13.32,3. En nuestro caso, la niña era portadora, además, de la variante polimorfa 9 qh++, que no tiene repercusión fenotípica. En caso de confirmarse el diagnóstico de SMD es imprescindible realizar un cariotipo a los padres, ya que en un 20% de los casos son portadores de alguna anomalía cromosómica balanceada2,3. En la serie de Fong, el 40% de los casos eran familiares3. En estas familias el riesgo de recurrencia es del 25%1,3.
A pesar de los avances en las técnicas diagnósticas, muchos fetos con malformaciones del desarrollo cortical escapan al diagnóstico prenatal, probablemente debido a: el desarrollo relativamente tardío del cerebro durante la gestación, cuando la ecografía no es realizada de forma rutinaria; limitaciones técnicas que impiden un detallado estudio del cerebro fetal; el limitado uso de la RM prenatal, que es la técnica de elección para el diagnóstico de estos trastornos y un incompleto conocimiento del desarrollo prenatal del córtex8.
Aunque Toi y otros autores2,4,8 no recomiendan una evaluación completa de las cisuras cerebrales como parte del examen anatómico fetal de rutina, pensamos que el conocimiento del patrón de sulcación puede ser muy útil y que las cisuras que aparecen más precozmente (parieto-occipital y calcarina) pueden ser fácilmente evaluadas cuando se visualizan los cuernos occipitales de los ventrículos laterales. No obstante, se precisan más estudios prospectivos para que la evaluación de estas estructuras forme parte del cribado prenatal8.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.