


Casos clínicos
Diagnóstico prenatal de la displasia campomélica. A propósito de un caso
A. B. Espejo
M. Marcelino
S. Franco
J. R. Herrero
C. Benito
B. González
M. Gallo
Hospital Materno Infantil de Málaga
Departamento de Obstetricia y Ginecología
Arroyo de los Ángeles, s/n
Málaga
Correspondencia:
Ana Belén Espejo Martínez
Acceso Río Adelfas. Urb. Sueño Azul, 27
29790 Benajarafe (Málaga)
Prenatal diagnosis of camptomelic dysplasia. A case report
Espejo AB, Marcelino M, Franco S, Herrero JR, Benito C, González B, Gallo M. Diagnóstico prenatal de la displasia campomélica. A propósito de un caso. Prog Obstet Ginecol 1999;42:251-255.
Fecha de recepción: 22/7/98
Aceptado para publicación 16/11/98
INTRODUCCIÓN
La displasia campomélica es una displasia esquelética infrecuente; con una incidencia aproximada de 1/150.000 nacimientos(1). Al igual que otras displasias es probable que incluya un grupo heterogéneo de trastornos, siendo el grado de severidad variable(2,3). Se ha considerado uniformemente como pronóstico fatal(1,4).
La displasia campomélica se asocia con mutaciones espontáneas o recurrentes a nivel del locux SOX9 situado en el brazo largo del cromosoma 17(5,6,7,8). Los estudios genéticos más recientes sugieren un patrón de herencia esporádico o autosómico dominante(4,6). Y se relaciona frecuentemente con inversiones sexuales autosómicas, con cariotipo 46, XY(2,4,5,6,7,8). Se caracteriza por una incurvación ventral de las tibias, fémures acortados, pie equino-varo severo, ausencia o hipoplasia de peroné, escápulas extremadamente pequeñas, tórax angosto e hipoplasia de los pedículos de las vértebras dorsales. Asimismo, suelen presentar: dilatación cerebroventricular, fisura palatina, cardiopatía congénita, escoliosis mediodorsal y pelvocaliectasias; produciéndose la muerte generalmente, por complicaciones secundarias a laringotraqueomalacias(1).
CASO APORTADO
Presentamos un caso de displasia campomélica cuyo diagnóstico de sospecha se realizó intraútero, en una mujer de 28 años, que acudió a la unidad de diagnóstico prenatal remitida por sospecha de displasia ósea esquelética, tras una exploración ecográfica rutinaria realizada en segundo trimestre. La paciente presentaba entre sus antecedentes personales, un hipotiroidismo en tratamiento sustitutivo con tiroxina. Entre sus antecedentes gineco-obstétricos, destaca la existencia de un aborto espontáneo (legrado).
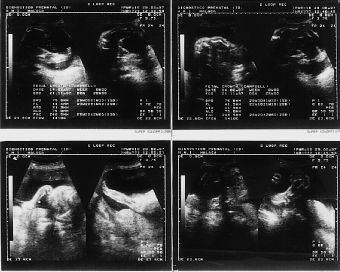
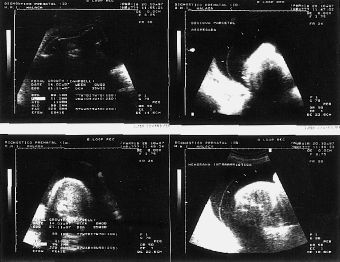
El examen ultrasonográfico, realizado en la Unidad de Diagnóstico Prenatal de nuestro hospital, en semana de amenorrea 28 demostró la existencia de un feto de sexo femenino con acortamiento de ambas tibias y peronés, con incurvación de fémures, con pies equinovaros y polihidramnios severo (Fig. 1). En controles ecográficos posteriores, se objetiva, un tórax angosto, un abdomen globuloso, con ausencia de cámara gástrica, importante acortamiento de fémures y la persistencia de un polihidramnios severo (Fig. 2) . En semana 31, se realiza el estudio citogenético del líquido amniótico, que nos revela un cariotipo masculino, 46 XY normal, y un nivel de alfafetoproteína en líquido amniótico de 2 mcg/ml.
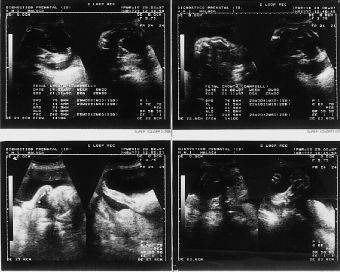
Figura 1.Examen ultrasonográfico en semana de amenorrea 28: feto de sexo femenino con acortamiento de ambas tibias y peronés, con incurvación de fémures, con pies equinovaros y polihibramnios severo.
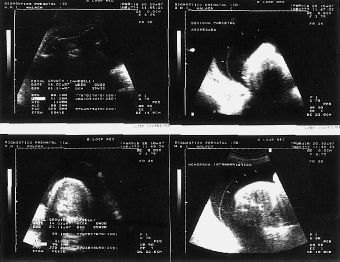
Figura 2.Control ecográfico en S.A. 35: un tórax angosto, un abdomen globuloso, con ausencia de cámara gástrica, importante acortamiento de fémures, con longitud correspondiente a S.A. 26 y la persistencia de un polihidramnios severo.
A la vista de los hallazgos ecográficos se nos plantea el diagnóstico el diagnóstico diferencial del cuadro con gran número de displasias óseas (tabla 1), focalizando nuestras sospechas hacia la displasia campomélica, por el hallazgo destacado de incurvación importante de fémur, tibia y peroné, que es corroborado en gran medida por el hallazgo en estudio citogenético de un cariotipo 46 XY; con la visualización en ecografía de genitales femeninos.
Tabla 1 | ||
| Tipo de displasia ósea | Características ecográficas | |
| Frecuentes | ||
| Displasia tanatofórica | Cráneo en hoja de trébol (14%); columna angosta; platispondilla. Epífisis femoral en «mango de teléfono». | |
| Acondrogénesis | +/ ausencia de cuerpo vertebral y osificación sacra. | |
| Osteogénesis imperfecta tipo II | Fracturas múltiples; hipomineralización de la calota craneal; huesos «gruesos». | |
| Raras | ||
| Hipofosfatasia congénita | Huesos delgados, delicados o ausentes; +/ fracturas; +/ disminución de la ecogenicidad ósea. | |
| Displasia campomélica | Incurvación anterior del fémur, la tibia y el peroné. | |
| Condrodisplasia punteada, tipo rizomélica | Epífisis punteadas; acortamiento rizomélica; contracturas; osificación dorsal y ventral de los cuerpos vertebrales. | |
| Acondroplasia homocigótica dominante | Se asemeja a la displasia tanatofórica (ambos padres afectos). | |
| Síndrome costillas cortas-polidactilia | Polidactilia; +/ fisura labial; quistes renales, microcefalia. | |
| Extraída de (1). | ||
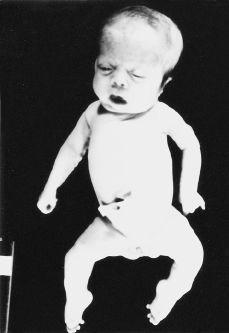
El embarazo curso con APP en semana de amenorrea 32, que requirió ingreso hospitalario para tratamiento tocolítico intravenoso. El parto se produjo de forma espontánea en semana de amenorrea 36, finalizando de forma eutócica. Obteniéndose un recién nacido fenotípicamente mujer (Fig. 3) 2,640 g de peso, con 38 cm de perímetro cefálico y 31 cm de perímetro torácico. El test de apgar al 1'', 5'' y 10'' fue: 3/5/7. Al tercer día de su nacimiento, el recién nacido evoluciona con un cuadro de insuficiencia cardíaca, produciéndose el exitus a los 23 días de vida.
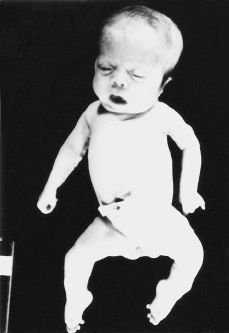
Figura 3.Recién nacido fenotípicamente mujer, con cráneo macrocéfalo-dolicocéfalo, fascies dismórficas con hipertelorismo, raíz nasal amplia; pabellones auriculares de implantación baja, tórax campaniforme; piernas arqueadas con pies zambos.
El examen radiológico postnatal mostró: macrocefalia-dolicocefalia, hipoplasia de vértebras cervicales. Ensanchamiento interpeduncular en columna lumbar; tórax campaniforme, con cierto grado de hipoplasia de la caja torácica; cardiomegalia con mínimo aumento de la vascularización pulmonar; escápulas hipoplásicas y elevadas; extremidades superiores de morfología normal; extremidades inferiores con arqueamiento a nivel de diáfisis femorales, con tibias y peronés hipoplásicos. Se observa una alteración global de la morfología y desarrollo de los huesos de los pies. El estudio cardiológico que objetiva la existencia de una CIV subaórtica de mediano tamaño con acabalgamiento aórtico. El estudio genético postnatal (estudio en sangre periférica, tras cultivo 72 horas, con técnicas de bandeo g y c), ratifica un cariotipo 46, XY.
El estudio anatomopatológico postmortem, nos revela: Macroscópicamente: cráneo macrocéfalo-dolicocéfalo, fascies dismórficas con hipertelorismo, raíz nasal amplia; pabellones auriculares de implantación baja, tórax campaniforme; piernas arqueadas con pies zambos; CIV subaórtica de 0,4 cm; riñones en herradura; vagina, útero, trompas de Falopio y gónadas visibles. El estudio microscópico nos muestra: gónadas: testículos disgenésicos con cordones seminíferos rudimentarios. Sistema esquelético con placas de crecimiento retardado y desorganizado; se informa como: Displasia campomélica con disgenesia gonadal.
COMENTARIOS
Las anomalías musculoesqueléticas fetales no son raras, siendo el papel del ecografista crucial en el diagnóstico prenatal y manejo de un embarazo afecto.
Sin embargo, el diagnóstico prenatal de las displasias óseas, pertenece a un apartado del diagnóstico prenatal bastante complejo(1,9). La dificultad diagnóstica estriba en que la variedad de los síndromes descritos y sus subtipos es mucho mayor que el conjunto de signos ecográficos existentes; por lo que el valor predictivo de cada uno de los signos ecográficos de forma aislada es muy baja(1).
No obstante, el descubrimiento fortuito de una anomalía de extremidades en una ecografía realizada por indicación obstétrica obliga a una búsqueda cuidadosa de otras características, que cuando se presentan, pueden permitir el diagnóstico prenatal confiable de un síndrome específico. El papel del ecografista, es pues, caracterizar la anomalía y buscar anomalías asociadas en un intento por determinar un diagnóstico específico prenatal, facilitando una información pronóstica esencial para el manejo obstétrico y posterior consejo genético.
En nuestro caso el diagnóstico se sospecha de displasia campomélica fue realizado ecográficamente, por el hallazgo de un arqueamiento típico característico de los huesos tubulares de las extremidades inferiores; siendo apoyado por el hallazgo de una discordancia entre el sexo ecográfico y el sexo genético revelado en el estudio citogenético. Señalando que el estudio de confirmación viene dado por el estudio radiológico postnatal y el examen anatomopatológico postmortem.
De cara al consejo genético posterior, señalar que numerosos estudios han puesto de manifiesto, la responsabilidad de mutaciones a nivel del gen SOX9 situado en el brazo largo del cromosoma 17(5,6,7,8), en el desarrollo de displasia campomélica e inversiones sexuales. Siendo el patrón de herencia esporádico o autosómico dominante(4,6).
Se confirma en este caso, el pronóstico fatal de este tipo de displasia, que se incluye en el grupo de displasias esqueléticas letales.
BIBLIOGRAFIA
1 Mahony BS. Evaluación ecográfica del sistema musculoesquelética fetal. En: Callen PW, editor. Ecografía en obstetricia y ginecología 3.ª ed. Argentina: De. Médica Panamericana; 1997. p. 284.
2 Schaler AJ, Foster JW, Kwok C y cols. Campomelic dysplasia with XY sex reversal: diverse phenotypes resulting from mutations in a single gene. Annals of the New York Academy of Sciences 1996;78:137-49.
3 Glass RB, Rosenbaum KN. Acampomelic dysplasia: further radiographic variations. Am J Med Genet 1997;69:29-32.
4 Mansour S, Hall CM, Pembrey ME, Youbg ID. A clinical and genetic study of Campomelic dysplasia. J Med Genet 1995;32: 415-20.
5 Meyer J, Sudbeck P, Held M y cols. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Human Molecular Genetics 1997;6:91-8.
6 Cameron FJ, Hageman RM, Cooke-Yareborough C y cols. A novel germ line mutation in SOX9 causes familial campomelic dysplasia and sex reversal. Human Molecular Genetics 1996;5:1625-30.
7 Wirth J, Wagner T, Meyer J y cols. Translocation breakpoints in three patients with campomelic dysplasia and autosomal sex reversal map more than 130 kb from SOX9. Human Genetics 1996;97:186-93.
8 Kwok C, Weller PA, Giolo S y cols. Mutations in SOX9, the gen responsible for Campomelic dysplasia and autosomal sex reversal. Am J Hum Genet 1995;57:1028-36.
9 Norgard m, Yankowitz J, Rhead W, Kanis AB, Hall BD. Prenatal ultrasound findings in hydrolethalus: continuing difficulties in diagnosis. Prenatal Diagnosis 1996;16:173-9.