


2ª PONENCIA
FACTORES DE RIESGO EN GINECOLOGÍA ONCOLÓGICA
Factores de riesgo en el cáncer de mama
F. Calero Cuerda
Jefe de Servicio de Ginecología Oncológica
Hospital Materno-Infantil La Paz
Profesor Asociado de la Universidad Autónoma de Madrid
Correspondencia:
Francisco Calero Cuerda
Sanchidrián, 38
Pozuelo de Alarcón
28224 Madrid
RESUMEN
Tras una exposición de los factores de riesgo más importantes en el cáncer de mama se establecen dos grupos de riesgo: alto y moderado. Las familias de alto riesgo tienen un gen de susceptibilidad al cáncer de mama con predisposición al cáncer de mama hereditario, donde se hace una amplia exposición del estado actual del arte. Las pacientes de riesgo moderado se seleccionan por los métodos de Claus y de Gail, justificando una cuantificación del riesgo. Se termina con las recomendaciones de prevención, vigilancia y de tratamiento de las pacientes y familias de alto riesgo.
INTRODUCCION
Desde hace tiempo se conocen los numerosos factores que modifican el riesgo de cáncer de mama, entre estos factores se incluyen: edad, edad temprana de la menarquia, edad tardía de la menopausia, nuliparidad, edad tardía para el primer embarazo, obesidad, exposición a altas dosis de radioterapia, ausencia de lactancia materna, antecedentes de algunos tipos de enfermedades benignas de la mama, antecedentes de carcinoma lobulillar in situ, antecedente de cáncer de mama e historia familiar de cáncer de mama en uno o más familiares de primer grado(1,2). Los factores relacionados con el estilo de vida, como la dieta elevada en grasas animales, consumo de alcohol y la actividad física también afectan al riesgo de cáncer de mama, aunque estos parámetros no muestren incremento elevado(1-3). Se considera pues que la mayoría de los factores de riesgo conocidos para cáncer de mama no son muy fuertes, aunque algunos de ellos adquieren mayor importancia, como son la edad, antecedentes familiares de cáncer de mama, historia personal de cáncer de mama y la presencia de enfermedad benigna de la mama confirmada con biopsia. Cuando se estudia por separado cada factor de riesgo se confirma que los antecedentes familiares de cáncer de mama y su patrón específico es el más importante, a tal punto que la mujer con algún familiar de primer grado afectado con cáncer de mama, el riesgo de presentar la enfermedad se incrementa dos a tres veces más en comparación con la mujer sin este antecedente(1,4-7).
FACTORES DE RIESGO
La etiología del cáncer de mama no es conocida, en numerosos estudios se investigan los posibles factores de riesgo para predecir y cuantificar el riesgo de padecer la enfermedad, aunque en más del 80% de mujeres con cáncer, éste aparece de manera esporádica sin factores de riesgo que lo explique. Comentamos los factores más conocidos, pero antes conviene fijar dos conceptos a los que repetidamente nos referiremos a continuación, uno es el riesgo relativo que resulta de la comparación de la proporción de afectación por la enfermedad en una población con la proporción de mujeres no expuestas en grupos similares de población; otro es el riesgo absoluto que resulta una probabilidad estadística, es decir la probabilidad de desarrollar cáncer de mama durante un intervalo de tiempo especificado, que se suele expresar como riesgo acumulado por la suma del riesgo anual a los 10 años o de por vida.
Sexo
Las mujeres tienen una mayor proporción de cáncer de mama que los hombres. Se conoce que el 99% de cánceres de mama aparecen en mujeres.
Edad
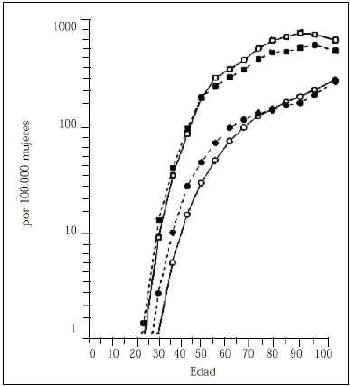
Es el factor de riesgo más importante. El riesgo de cáncer de mama aumenta rápidamente con la edad durante los años de actividad sexual en la mujer, después de la menopausia la frecuencia se incrementa pero con menos rapidez (Fig. 1). Del estudio a gran escala efectuado en el programa Surveillance, Epidemiology and End Results (SEER) de Estados Unidos(8) se desprende la probabilidad de padecer la enfermedad y de morir por esta causa con intervalos distintos de edad. En los resultados que se exponen en la figura 1 resaltan dos hechos importantes, la probabilidad de padecer cáncer de mama muestra íntima relación con el incremento de la edad, y la incidencia de la enfermedad es dos a tres veces mayor que la probabilidad de morir de cáncer de mama, a título de ejemplo la probabilidad de muerte a los 65 años es similar a la incidencia de la enfermedad a los 45 años.

Figura 1.Frecuencia y mortalidad del cáncer de mama según la edad de la paciente(8).
Antecedentes personales de cáncer de mama
Las pacientes tratadas por un tumor primario de mama tienen un riesgo de desarrollar un segundo cáncer de mama de tres a cinco veces más alto que la población general(9). Dentro de estas pacientes hay un subgrupo de mujeres en que la probabilidad es aún mayor, me refiero a las pacientes tratadas en edad joven, de menos de 50 años con esperanza de vida de más de 20 años con carcinoma ductal in situ y tipos histológicos de buen pronóstico.
También presentan mayor riesgo de cáncer de mama contralateral las mujeres con antecedentes familiares de cáncer de mama y las que tuvieron el tumor primario multicéntrico.
Factores hormonales y reproductivos
La duración total de la actividad hormonal del ovario es el condicionante del riesgo de padecer cáncer de mama y por ello la fisiología hormonal endógena juega un papel crucial(10). La importancia que desempeñan los estrógenos y los gestágenos no está suficientemente aclarada, pero se estima aumento del riesgo de padecer cáncer de mama en las situaciones con mayor intervalo de tiempo en que la mama está sometida al influjo hormonal del ovario como sucede en las dos situaciones de menarquia precoz y menopausia tardía(11,12).
El intervalo de tiempo transcurrido entre la telarquia y el primer embarazo a término se muestra un período crítico para determinar el riesgo de cáncer de mama(13). Con el inicio de la pubertad las hormonas segregadas por el ovario inducen la proliferación del epitelio ductal de la mama, es decir, que los estrógenos y progesterona estimulan la actividad mitótica de las células de los conductos terminales de la mama(14). En las sociedades que la menarquia es temprana probablemente debida a una dieta conveniente, la producción de estrógenos en la adolescencia es más prolongada, como la secreción de estrógenos condiciona la proliferación del epitelio de la mama, el proceso transcurre de forma continua hasta el final del primer embarazo, de tal manera que si el intervalo se acorta la incidencia de cáncer de mama disminuye y en la menarquia precoz el riesgo de cáncer de mama se ve incrementado. Una mujer con menarquia antes de los 12 años y con instauración cíclica de la menstruación tiene un aumento del riesgo de 50% comparado con las mujeres que tienen la menarquia después de los 15 años(15). Aproximadamente existe un 15% de disminución del riesgo por cada año que se retrasa la menarquia.
Durante la gestación las hormonas esteroideas se segregan en cantidad considerable y contribuyen a la diferenciación de las células de los lobulillos, que se preparan para la secreción láctea, lo que les hace resistentes a la carcinogénesis. Las mujeres que tienen hijos disminuye el riesgo de cáncer de mama en comparación con las nulíparas, pero el efecto protector del embarazo a término se cumple hasta la edad de los 30 años, porque en la mujer con primer embarazo después de los 35 años el riesgo relativo aumenta y con incremento progresivo con la edad de la primera gestación. En algún estudio(16) el efecto protector de la multiparidad se limita a las mujeres en que el cáncer se diagnostica después de los 50 años, pero antes de esa edad la nuliparidad no significa un mayor riesgo de cáncer de mama.
La edad del primer embarazo a término es un factor importante de riesgo, demostrado por MacMahon y cols. en el estudio internacional(17). Las mujeres con primer embarazo a término después de los 30 años tienen doble de riesgo que las que tienen el primer embarazo antes de los 18 años. Las nulíparas tienen riesgo similar a la mujer con primer embarazo a los 30 años.
Se piensa que la lactancia materna disminuye la incidencia del cáncer de mama, no obstante los estudios epidemiológicos han sido incapaces de estimar de manera precisa el efecto de la lactancia sobre el riesgo de cáncer de mama. El efecto protector se limita a las mujeres con tumores diagnosticados en el período de la premenopausia(18) y desde luego con el condicionante de que el período de lactancia sea prolongado(19), como se desprende del estudio caso control efectuado en las mujeres chinas donde la lactancia es prolongada como norma con lo que el riesgo de cáncer de mama disminuye conforme aumentan los años de lactancia(20). El aborto inducido parece que empeora el riesgo(21) aunque no está absolutamente establecido.
La edad tardía de la menopausia se relaciona con aumento del riesgo de cáncer de mama. Las mujeres con menopausia natural antes de los 45 años tienen la mitad de riesgo de cáncer de mama comparado con las mujeres que tienen la menopausia después de los 55 años(15,22). La menopausia artificial quirúrgica o con radiaciones tiene un efecto protector cuando sucede de manera precoz. El efecto protector de la castración se relaciona directamente con la edad en que se realiza, es máxima si acaece antes de 35 años, lo que confiere un 64% de protección y conforme aumenta la edad en que se hace la castración disminuye la protección(15). El efecto protector del riesgo no empieza a manifestarse hasta pasados los cinco primeros años de la menopausia, llegando a un 35% de protección a los 10 años y hasta un 50% a los 20 años.
Distribución geográfica
El carcinoma de mama es más frecuente en Estados Unidos, Canadá y países de Europa septentrional y se le observa con menos frecuencia en Asia, América Latina y África. Las diferencias se explican por factores genéticos y ambientales, fundamentados principalmente en estudios de mujeres emigrantes, por ejemplo las mujeres japonesas que viven en Hawai tienen doble de riesgo que las que viven en Japón. De la misma manera las mujeres chinas que viven en California tienen más del doble de riesgo que las mujeres que viven en China(23,24). En ambas poblaciones de emigrantes a Estados Unidos aumenta progresivamente la incidencia de cáncer de mama en las generaciones sucesivas que llega casi a igualarse con las mujeres norteamericanas.
Las mujeres de nivel socioeconómico alto estimado por el nivel de educación e ingresos económicos tienen mayor riesgo, aunque la tasa de mortalidad sea inferior(25). También se observa mayor incidencia en las mujeres que viven en la ciudad que en zonas rurales.
Estilo de vida
La relación entre el peso y el cáncer de mama depende estrictamente de la edad. Para las mujeres postmenopáusicas y con edad de 60 años el riesgo se incrementa con el aumento del peso, se estima que un exceso de 10 kg puede aumentar tanto el riesgo como en un 80%(26). El aumento del riesgo posiblemente se debe a la producción de estrógenos por la grasa a partir de los andrógenos suprarrenales. En contraste en las mujeres premenopáusicas el exceso de peso muestra resultados contradictorios. Por los datos disponibles que proceden de estudios experimentales en animales, de los estudios ecológicos, de las poblaciones migratorias y de los ensayos caso control avalan la asociación entre consumo de grasas y cáncer de mama. Sin embargo los estudios de cohorte no confirman esta asociación, probablemente debido a la escasa variabilidad de la ingesta. El efecto de los diferentes tipos de grasa no está aclarado, hasta el punto de considerar algunos tipos de ácidos grasos como posibles quimioprotectores. Durante mucho tiempo se estima que el aumento de grasas de origen animal, sobre todo de grasas saturadas y los derivados lácteos, supone un factor de riesgo importante en el cáncer de mama. pero no existe acuerdo unánime y los estudios apuntan más a que el riesgo se debe a la ingesta de calorías totales que al solo consumo de grasa animal. En los estudios de correlación existe una asociación lineal entre el consumo de grasa, dieta hipercalórica y cáncer de mama, aunque esto no se cumple en los estudios caso control, ni en los prospectivos. Las personas vegetarianas y con hábito de vida de reducción del consumo de grasa se demuestra una protección frente al cáncer de mama. En la actualidad se atribuye una explicación adicional a la obesidad y aumento excesivo de peso, compatible con las posibles alteraciones en la secreción y metabolismo de las hormonas esteroideas, y quizá este mecanismo sea la explicación del aumento de riesgo de cáncer de mama.
Los estudios epidemiológicos sobre la alimentación con aceite y proteínas de soja que contiene fitoestrógenos están de actualidad, sabemos que estos compuestos se unen a los receptores plasmáticos de estrógenos y actúan como estrógenos pero con acción más débil. En las mujeres con dieta rica en proteínas de soja se demuestran excreción acrecentada de isoflavonoides y se estima como posible mecanismo el que estas mujeres así alimentadas tienen una más baja incidencia de cáncer de mama. De la misma manera en el estudio prospectivo(27) de caso control sobre fitoestrógenos y cáncer de mama se observa una reducción evidente de la incidencia de cáncer de mama entre mujeres con una ingesta rica en proteínas de soja, medida por la excreción de fitoestrógenos.
En muchos estudios caso control se demuestra la asociación entre talla y riesgo de cáncer de mama. De la misma manera en los estudios de cohorte verificados en los países escandinavos se demuestra la asociación del riesgo incrementado de 1,1 para aumento de 5 cm en la talla y de 2 para más de 8 cm de incremento(28,29).
El alcohol es el componente de la dieta para el que existe una relación evidente con aumento del riesgo de cáncer de mama, así en la revisión de los 17 estudios de cohorte y de caso control publicados, se detecta en 14 de ellos una relación cierta(30). El aumento del riesgo se demuestra en pacientes premenopáusicas que consumen más de 15 gramos de alcohol al día y el riesgo se incrementa en las mujeres que consumen más de esa cantidad.
No existe relación con el hábito del tabaco, consumo de cafeína o metilxantinas, ni con el estrés, ni con la depresión.
El ejercicio físico moderado reduce la incidencia de cáncer de mama y se estima que el ejercicio intenso de las atletas en la juventud protege del desarrollo de tumores malignos en la mama(31,32). Diversos mecanismos todos ellos de índole hormonal están imbricados, de una parte es evidente la reducción del número de ciclos ovulatorios, también se reduce el peso, no hay obesidad ni depósito de grasa con lo que disminuye el aporte de tejido graso para la aromatización de andrógenos a estrógenos(33).
Cirugía mamaria previa
Desde hace tiempo se estima la existencia de una relación entre enfermedades benignas de la mama y el riesgo de cáncer. Se debe a los estudios de Dupont y Page(34) y posteriormente a la contribución de los patólogos del Colegio Americano la clasificación en tres categorías de lesiones de la mama para relacionarlas con el riesgo. Las lesiones con histología de hiperplasia ductal leve o lesiones no proliferativas, que comprenden la mayoría de las muestras de biopsia no se observa incremento del riesgo. En la enfermedad proliferativa o hiperplasia ductal sin atipias aumenta el riesgo de cáncer de mama en 1,5 a 2 veces. En la hiperplasia ductal atípica el riesgo relativo de padecer cáncer de mama se incrementa cinco veces y en las pacientes diagnosticadas de carcinoma lobulillar in situ el riesgo relativo se incrementa 8 a 10 veces. De la misma manera en el estudio prospectivo(35) de 121.700 pacientes con enfermedad benigna de la mama sin lesión proliferativa el riesgo relativo de cáncer de mama es de 1,6 pero se eleva el riesgo a 3,7 cuando la histología de la biopsia es de hiperplasia atípica. El riesgo se acumula en las mujeres premenopáusicas y se acentúa cuando además existen antecedentes familiares de cáncer de mama.
Por otra parte, independientemente del resultado patológico de la muestra de biopsia de mama se estima que una sola intervención dobla el riesgo de cáncer de mama y si hay más de una biopsia se aumenta el riesgo cinco veces al de la población general.
Tratamiento hormonal
Existe una gran preocupación respecto del tratamiento hormonal sustitutivo y la posibilidad del aumento de riesgo de desarrollar cáncer de mama(36-38). Los estudios de cohorte, caso control y meta-análisis parece concluir que con tratamientos de larga duración, de más de cinco años aumenta ligeramente el riesgo de padecer cáncer de mama. Se describe aumento del riesgo relativo de 1,3 en mujeres con tratamientos muy prolongados, de más de 10-20 años.
La toma de anticonceptivos orales y su relación con el cáncer de mama ha sido objeto de muchos estudios, pero por los resultados de estudios amplios no se conoce que haya aumento de incidencia(39,40). La discreta elevación del riesgo del amplio estudio epidemiológico efectuado por la SEER(41) y en la revisión de la literatura entre las mujeres consumidoras de anticonceptivos orales(42) lo relacionan con una duración prolongada de la toma mayor de diez años, con el inicio de manera precoz, antes de transcurridos cinco años de la menarquia y al empleo de anticonceptivos de gran potencia gestágena. De la misma manera se describe aumento del riesgo de cáncer de mama(43) en las mujeres que toman la medicación durante muchos años y no han tenido embarazos.
Exposición a radiaciones ionizantes
La exposición a radiaciones ionizantes aumenta el riesgo de cáncer de mama como se demuestra en los estudios sobre mujeres supervivientes en las explosiones atómicas de Hiroshima y Nagasaki, y de las mujeres con exposición a exploraciones radiográficas repetidas. Está bien establecido que las radiaciones ionizantes en dosis moderada o alta antes de los 40 años aumenta el riesgo de cáncer de mama y el riesgo se incrementa con dosis más alta(44). También se describe mayor proporción de cáncer de mama en mujeres tratadas con radioterapia por enfermedad de Hodgkin(45). El riesgo se acumula en mujeres jóvenes entre 10 y 20 años y es escaso en las mujeres con más de 35 años. El riesgo de cáncer de mama se hace evidente hacia los cinco y diez años después de la radiación, con el pico de máxima incidencia a los 15 años y con una mayor mortalidad a los 20 años. Este incremento del riesgo se mantiene durante toda la vida.
Antecedentes familiares de cáncer de mama
Se admite que existen dos tipos de presentación del cáncer de mama: familiar y esporádico. De una manera empírica se define como cáncer de mama familiar a aquel que se presenta en dos o más individuos de un árbol genealógico que abarca al menos tres generaciones, mientras que la definición de cáncer de mama hereditario se circunscribe a aquellas familias con un gen de susceptibilidad al cáncer y un desarrollo genealógico autósomico dominante.
En la tabla 1 se expone el incremento del riesgo relativo de cáncer de mama en mujeres con un familiar de primer grado afectado, cuyo índice de riesgo oscila entre 1,5 a 2(14) y hasta de nueve cuando la hermana o la madre tienen la enfermedad en la premenopausia y la afectación es bilateral(46,47).
Tabla 1 Riesgo relativo de padecer cáncer de mama según el antecedente familiar de primer grado (madre o hermana)(46,47) | |
| Características del diagnóstico | Riesgo relativo |
| Premenopausia | 3,0 |
| Cáncer bilateral de mama | 5,0 |
| Premenopausia y bilateral | 9,0 |
| Postmenopausia | 1,5 |
En el 15% de los casos de cáncer de mama existe una relación familiar que no se explica solo por factores ambientales y en el 5-7% la herencia es el factor determinante. En España conocemos el resultado del estudio caso control(48) por el que la historia familiar está presente en el 18,5% de los casos de enfermedad, y la relación es de primer grado en el 10,6% frente al 2,8% del grupo control..
BASES PARA EL CALCULO DE RIESGO
La valoración del riesgo individual empieza por hacer una historia detallada de la paciente y de sus antecedentes familiares, considerando la presencia de cáncer de mama en líneas materna y paterna, para si es necesario construir un árbol genealógico(49). Como una guía para el entendimiento clínico y las recomendaciones a las mujeres con historia familiar de cáncer de mama, Hoskins y cols.(50) estratifican las mujeres con historia familiar de cáncer de mama en dos grupos, aquellas con un riesgo moderado y aquellas con un riesgo elevado.
Las mujeres de familias con alto riesgo tienen una predisposición genética con una mutación de los genes que confieren susceptibilidad a ciertos tipos de cáncer o síndromes asociados. Las formas hereditarias de cáncer se caracterizan por diagnóstico del cáncer de mama en edad joven, enfermedad bilateral y tumores multifocales. Los antecedentes familiares próximos de cáncer de mama u ovario confieren riesgo elevado aunque sea verdad que algunas familias son demasiado pequeñas para demostrar el riesgo elevado de forma aparente y sean clasificadas de manera errónea como de riesgo moderado. Las mujeres con historia familiar cargada, consideradas de alto riesgo corresponden aproximadamente al 5% del conjunto de pacientes con cáncer de mama y se aproxima al 25% de las enfermas con diagnóstico efectuado antes de los 30 años de edad(51). La mayor parte de pacientes de este pequeño número de enfermas con cáncer de mama de alto riesgo, en los estudios publicados hasta la fecha, presentan tres regiones del genoma implicadas en la predisposición al cáncer de mama. El gen BRCA1 localizado en la región 17q, el gen BRCA2 localizado en el cromosoma 13q12-13, ambos asociados al tipo familiar, y el p53 que es el gen implicado en el síndrome de Li-Fraumeni. Otro pequeño número de familias de alto riesgo presentan síndromes hereditarios distintos que se relacionan con un riesgo elevado de cáncer de mama, que por lo regular es posible identificarlas por los cuadros clínicos que les acompañan.
Cuando se identifican los marcadores moleculares de riesgo en individuos de una familia, el riesgo de cáncer de mama a parte de ser elevado se mantiene de por vida, con lo que es posible definir con gran precisión a miembros de la familia con riesgo próximo al 100%, o al contrario mujeres con riesgo comparable a la población general. Consideramos que una vez detectadas las familias de alto riesgo se las debe referir a centros que proporcionen acceso a estudios genéticos, porque tales familias son candidatas para plantear estrategias de prevención, tratamiento y registro.
En el análisis inicial de riesgo de cáncer de mama se utiliza sobre todo los antecedentes familiares de cáncer(52,53) sin incorporar otros factores, a esta conclusión se llega por los resultados del estudio comparativo de las 4.730 mujeres diagnosticadas histológicamente de cáncer de mama en el programa Cancer and Steroid Hormone (CASH) con el grupo control de 4.688 mujeres de la misma edad y área geográfica. La información de los antecedentes familiares se realiza en la entrevista personal cuando se hace la mamografía. Los resultados muestran en el grupo diagnosticado de cáncer de mama que el 11% de las mujeres tienen un familiar de primer grado afectado, comparado con el 5% del grupo control(54), pero en menos del 1% de pacientes tienen en los antecedentes dos miembros afectados (madre y hermana). A este simple esquema se le conoce hoy día como el modelo Claus o modelo CASH, en que el punto esencial es el tener al menos un miembro de primer grado de la familia afectado y, por tanto, se considera un modelo genético. El efecto de demostrar mutaciones en las familias seleccionadas está en función de la edad, se estima que el 36% de los casos de cáncer de mama en mujeres con edades entre 20 y 29 años se atribuye a un gen de susceptibilidad al cáncer con herencia autosómica dominante, que conforme aumenta la edad disminuye la proporción hasta el 1% alrededor de los 80 años.
Por lo general, las familias con un riesgo moderado de cáncer de mama manifiestan la presencia de antecedentes de cáncer de mama en uno o dos miembros de la familia en segundo grado, a menudo el diagnóstico se hace en la postmenopausia, en ausencia de cáncer de ovario u otro síndrome familiar definido (Tabla 2). Estas familias constituyen un grupo heterogéneo, donde quizá se incluyen factores esporádicos o exposiciones ambientales como el estilo de vida más que con casos verdaderamente hereditarios. Se han desarrollado varios modelos para valorar el riesgo individual, que son de utilidad para hacer recomendaciones a los familiares con riesgo moderado(54-57). El modelo Gail(55) se basa en los datos del Breast Cancer Detection Demonstration Project (BCDDP) que reunió en un programa de detección precoz a 280.000 mujeres en 28 centros hospitalarios estadounidenses a mediados de los años 70(58,59). A partir de la información de los 2.852 nuevos cánceres de mama diagnosticados en los cinco años de programa de mamografía anual se perfecciona un modelo de riesgo que incorpora siete factores: sexo, raza, edad, edad de la menarquia, edad del primer parto a término, número de biopsias mamarias y número de parientes de primer grado con cáncer de mama. Después de una segunda consideración se perfecciona un modelo final limitado a mujeres blancas para el cálculo del riesgo basado en la edad de la menarquia, edad del primer parto, el número de parientes de primer grado afectados y el número de biopsias mamarias.
Tabla 2 Riesgo moderado de cáncer de mama para los familiares | ||
| Familiar afectado. Número y parentesco | Edad del familiar afectado. Años | Riesgo acumulado de cáncer de mama estimado hasta los 80 años (%) |
| 1. En primer grado | < 50 | 13-21 |
| 1. En primer grado | >= 50 | 9-11 |
| 1. En segundo grado | < 50 | 10-14 |
| 1. En segundo grado | >= 50 | 8-9 |
| 2. En primer grado | < 50 | 35-48 |
| 2. En primer grado | >= 50 | 11-24 |
| 2. En segundo grado | < 50 | 21-26 |
| 2. En segundo grado | >= 50 | 9-16 |
| Adaptado de Claus(54). | ||
El modelo Gail produce un cálculo preciso y no supone ningún modelo genético, que se utiliza en el asesoramiento clínico de mujeres preocupadas, se ha validado dos veces(60,61) y adaptado para identificar mujeres con riesgo elegibles para la inclusión en el estudio de quimioprevención con tamoxifeno(62).
FAMILIAS CON RIESGO AUMENTADO DE CANCER DE MAMA
El modelo Claus de análisis de riesgo empírico, está diseñado para mujeres con alto riesgo de padecer cáncer de mama por sus antecedentes familiares. Este modelo supone que la susceptibilidad al cáncer de mama se transmite genéticamente como rasgo autosómico dominante. Los cálculos de riesgo se presentan en forma tabular y toman en cuenta la edad de diagnóstico de la neoplasia maligna en el miembro de la familia y el grado de relación del afectado con la paciente que consulta. El modelo Claus considera los parientes de primer grado y también en ciertos casos, incorpora parientes de segundo grado tanto maternos como paternos.
Estudios epidemiológicos múltiples demuestran que los antecedentes familiares de cáncer de mama es un factor predictivo para padecer cáncer de mama(4-7, 63). El riesgo se incrementa dos a tres veces de tener un antecedente familiar de primer grado a no tenerlo. El cáncer de mama bilateral es un hecho clínico que se asocia a una susceptibilidad hereditaria a padecer cáncer de mama, siendo este tipo de patología de dos a tres veces más frecuente en familias de alto riesgo(64). Una característica epidemiológica importante es la asociación fuerte entre cáncer de mama familiar y cáncer de ovario. La asociación en una mujer de cáncer de mama y ovario como neoplasias primarias independientes sugiere la existencia de factores de riesgo compartidos entre estas dos enfermedades(65) y se observa incremento de cáncer de ovario de 1,5 a 1,9 veces entre miembros de estas familias(66,67). Otro hecho importante a tener en cuenta en el cáncer de mama familiar es la relación inversa entre el riesgo relativo de los familiares y edad a la cual se hace el diagnóstico de cáncer de mama en el familiar, siendo éste de 4,3 si el cáncer se diagnostica a los 30 años, de 2,7 si se diagnostica a los 40 y de 1,7 si se hace a los 50. Estos riesgos relativos aumentan si hay varios miembros afectados.
La aptitud para determinar mutaciones de los genes de susceptibilidad: BRCA1 y BRCA2 en algunos laboratorios permite la definición de una nueva subpoblación de mujeres con riesgo incrementado de desarrollar cáncer de mama y de ovario. Una vez diagnosticada esta susceptibilidad genética, se suscitan controversias en las recomendaciones del seguimiento y la prevención del cáncer en estas mujeres(68). Los análisis moleculares para el diagnóstico del riesgo individual de cáncer de mama todavía no están disponibles como uso rutinario, por otra parte cuando se disponga del estudio genético se presentarán dilemas, sobre todo desde los puntos de vista ético y terapéutico.
SUSCEPTIBILIDAD GENÉTICA AL CANCER DE MAMA: BRCA1 Y BRCA2
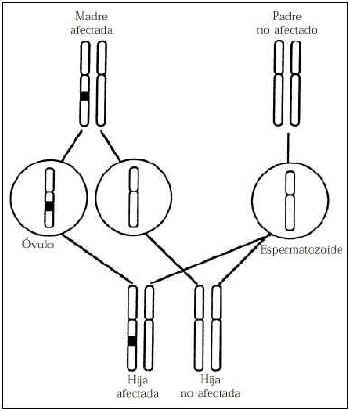
Se identifican dos genes de susceptibilidad de cáncer de mama denominados BRCA1 y BRCA2(69-73), y un tercero (BRCA3) se presume su existencia con bastante verosimilitud(74) (Tabla 3). En las familias portadoras de mutaciones de los genes de susceptibilidad de cáncer de mama se hereda de forma autosómica dominante de tal manera que se afectan todos los individuos de cada generación que heredan el mencionado gen (Fig. 2). Durante la gametogénesis se constituyen las células germinales que portan sólo un miembro de un par de cromosomas. En este ejemplo los espermatozoides son iguales porque el padre no está afectado. En la génesis del óvulo la mitad de las células germinales son portadoras de la mutación y la otra mitad no. En el caso de herencia dominante todos los hijos que llevan el cromosoma afectado resultan afectados (100% de penetración). En el caso de la herencia recesiva los hijos deben heredar dos copias de la mutación, uno de cada progenitor, padre y madre afectados para afectarse. Por ello a los dos genes se les considera que actúan como genes de alta penetración y la mayoría de las mujeres portadoras desarrollan cáncer de mama o de ovario a lo largo de su vida.

Figura 2.Herencia autosómica dominante. Se muestra un par de cromosomas de cada progenitor.
Tabla 3 Genes implicados en la susceptibilidad al cáncer de mama | ||
| Gen | Cromosoma | Manifestación clínica |
| BRCA1 | 17q21 | Síndrome hereditario familiar de mama y ovario |
| BRCA2 | 13q12-13 | Síndrome hereditario familiar de mama y ovario |
| BRCA3? | 8p12-22? | ? |
El estudio y aislamiento de los genes BRCA1 y BRCA2(71-75) han extendido nuestros conocimientos sobre la herencia del cáncer de mama. La identificación del locus genético en el cromosoma 17q(69,70) relacionado con el cáncer de mama intensifica la investigación para identificar y secuenciar a los genes asociados con susceptibilidad para cáncer de mama. Cabe el mérito a Miki y cols.(71) por medio de clonación posicional la identificación del espectro de un gen grande 17q21 que tiene 5.592 nucleótidos distribuidos en unas 100.000 bases de ADN genómico, está compuesto de 22 exones codificantes y codifica una proteína de 1.863 aminoácidos.
El gen BRCA2 está localizado en el cromosoma 13q12-13(76) y clonado en 1995(72,73), las alteraciones de este gen están vinculadas al cáncer de mama masculino, siendo el riesgo de cáncer de ovario menor que el de las familias con BRCA1. El gen BRCA2 tiene 11.385 nucleótidos distribuidos en 70.000 bases de ADN genómico y 27 exones codificantes, que codifican una proteína de 3.418 aminoácidos(76).
Pero es verdad que hasta en un 30% de las familias de alto riesgo no es posible confirmar ni la mutación BRCA1 ni la unión a BRCA2, por lo que se estima que en estas familias están presentes otros factores genéticos de susceptibilidad al cáncer aún no identificados. Como ejemplo citamos a tres de las cuatro familias de Hungría con antecedentes de seis casos de cáncer de mama u ovario(77); en dos de seis familias con cáncer de mama en el varón y 15 de 23 familias en la serie estudiada por la Agencia Internacional de Investigación del Cáncer(74); en cuatro de 25 familias con antecedentes familiares de ambos cánceres de mama y ovario, con dos afectadas de cáncer de ovario(78) y nueve de 48 familias americanas con más de cuatro casos de cáncer de mama y ovario(79). Estas observaciones junto con los estudios de pérdida de heterocigosidad en familias con cáncer de mama, son las que permiten a los investigadores afirmar la posible existencia de otros genes, en particular el denominado BRCA3 y posiblemente localizado en el cromosoma 8(80).
Frecuencia de las mutaciones BRCA1 y BRCA2
La frecuencia de las mutaciones BRCA1 y BRCA2 en familias de alto riesgo de los países que publican resultados se resume en la tabla 4, donde las familias de alto riesgo vienen definidas porque en sus antecedentes hay tres familiares con cáncer de mama o al menos dos afectadas si hay un caso de cáncer de ovario o un cáncer de mama en varón.
Tabla 4 Familias y pacientes con síndrome familiar de cáncer de mama y ovario (familias con tres o más casos de cáncer de mama y/o ovario) de varios países investigadas para la herencia de mutaciones de los Genes BRCA1 y BRCA2 | |||
| Países | N.º de familias con mutaciones BRCA1 | N.º de familias estudiadas BRCA2 | Referencias |
| Rusia | 15/19 (79%) | | 81 |
| Israel | 16/34 (47%) | 8/34 (24%) | 82 |
| Canadá | 12/30 (40%) | 8/49 (16%) | 83, 84 |
| Estados Unidos | 69/179 (39%) | 24/94 (25%) | 74, 79, 85-92 |
| Italia | 21/73 (29%) | | 93-95 |
| Francia | 38/160 (24%) | 14/77 (18%) | 96, 97 |
| Suecia y Dinamarca | 24/106 (23%) | 12/106 (11%) | 78, 98 |
| Hungría | 7/32 (22%) | 4/32 (13%) | 77 |
| Gran Bretaña | 71/339 (21%) | 25/290 (9%) | 59, 99-101 |
| Alemania | 9/49 (18%) | | 102, 103 |
| Holanda y Bélgica | 71/517 (14%) | | 104, 105 |
| Noruega | 3/25 (12%) | - | 106 |
| Japón | 2/20 (10%) | | 107 |
| Islandia | 1/11 (9%) | 7/11 (64%) | 108 |
| Finlandia | | 8/100 (8%) | 109 |
Lo primero que llama la atención es que la proporción de familias de alto riesgo atribuibles a mutaciones del BRCA1 varía ampliamente de un país a otro. La mutación BRCA1 es con mucho muy frecuente en Rusia ya que aparece en el 79% de familias con cáncer de mama y ovario, le sigue en frecuencia Israel con el 47% de mutaciones. Las familias de alto riesgo de Canadá y Estados Unidos tienen una frecuencia intermedia porque están constituidas exclusivamente por emigrantes. Inmediatamente después Italia con el 29% de familias. Entre el 20 y 25% de las familias de alto riesgo de Francia, Países escandinavos, Hungría y Gran Bretaña tienen mutaciones del gen BRCA1. Una frecuencia menor del 20% sucede en otros países como son Alemania, Holanda y Bélgica, Noruega, Japón y en Islandia.
En todos los países a excepción de Islandia la frecuencia de BRCA1 es 1,5 a dos veces más alta que la mutación del gen BRCA2, se trata de explicar porque la prevalencia sea efectivamente más baja, o que la penetrancia sea menor o que la edad de aparición sea más tardía. No obstante se considera que la distinta prevalencia sea efectivamente real, como se comprueba en las familias de Estados Unidos en las que las mutaciones del BRCA1 es el doble de frecuentes que las del BRCA2(74,79,86,87). Ocurre al contrario solamente en Islandia que es más frecuente las mutaciones del gen BRCA2 que la del BRCA1, donde una mutación, la 999 del cinco es la única que explica todos los cánceres hereditarios de mama y ovario(108). Esta mutación se la observa exclusivamente en Islandia y en dos familias de Finlandia(78,106,109). La preponderancia de una mutación y precisamente del gen BRCA2 representa uno de los ejemplos de mutaciones de una población donde la proporción es baja y existe endogamia debido al aislamiento de la población(110).
Las familias que tienen en sus antecedentes un cáncer de mama en varón se exponen separadamente en la tabla 5, donde se observa que es más frecuente la mutación del BRCA2. Así se observa en las familias estudiadas de Hungría y de Islandia. En los datos combinados de varias publicaciones de Estados Unidos se demuestra que el gen BRCA2 es responsable del 90% de los cánceres de mama en el varón(74,90,111), pero con una considerable frecuencia menor del cáncer de mama en el varón(90,111), que representa el 19% de los casos estudiados.
Tabla 5 Determinaciones de mutaciones en familias con antecedentes de cáncer de mama en el varón | |||
| Países | N.º de familias con mutaciones BRCA1 | N.º de familias estudiadas BRCA2 | Referencias |
| Estados Unidos | 2/24 (8%) | 12/64 (19%) | 74, 90, 111 |
| Hungría | 0/6 (0%) | 2/6 (33%) | 77 |
| Islandia | 0/10 (0%) | 9/10 (90%) | 108 |
¿Cuántos cánceres de mama se deben al BRCA1 y BRCA2?
Varios grupos sobre modelos individuales y familiares estudian las mutaciones del BRCA1 y BRCA2(112-114). La tabla 6 reúne los casos no seleccionados por la historia familiar sino exclusivamente estudiados por tratarse de pacientes con carcinoma de mama. Se observa que entre el 6 y 10% de los cánceres de mama tienen mutaciones para el BRCA1 y/o BRCA2, a excepción de Israel donde la frecuencia es más alta, que llega al 15%(116). La proporción estimada también es dependiente de la edad de diagnóstico, así antes de los 40 años se observa una frecuencia del 8,2%, es del 4% para edades comprendidas entre 40 y 49 años y del 1% para aquellas entre edades de 50 y 70 años(120,121).
Tabla 6 Frecuencia de mutaciones BRCA1 y BRCA2 en pacientes con cáncer de mama sin seleccionar por la historia familiar | |||
| Países | N.º de familias con mutaciones BRCA1 | N.º de familias estudiadas BRCA2 | Referencias |
| Islandia | | 42/497 (8%) | 115 |
| Israel | 23/243 (9%) | 14/343 (6%) | 116 |
| Italia | 4/49 (8%) | | 95 |
| Japón | 8/179 (4%) | 2/103 (2%) | 117-119 |
Los miembros asociados en el consorcio internacional para cáncer de mama reúnen datos clínicos y de vínculo genético de 237 familias con cáncer de mama(122), elegidas porque tienen como antecedente cuatro o más casos de cáncer de mama diagnosticados, al menos un caso de cáncer de ovario o cáncer de mama en varón. Las determinaciones de las mutaciones en el gen BRCA1 se encuentran en 123 casos (52%), las del gen BRCA2 en 76 pacientes (32%), y no se encuentra mutación alguna en 38 pacientes (16%). Entre las familias que incluyen en los antecedentes algún varón con cáncer de mama se encuentra el gen BRCA2 en el 76% de los casos.
¿Todas las portadoras de BRCA1 y BRCA2 desarrollan cancer?
Se estima que en las mujeres portadoras de mutaciones BRCA1 y BRCA2 se desarrolla cáncer de mama en el 87% de los casos, considerado como hereditario y en el 40 a 60% de los casos se desarrolla cáncer de ovario(120). Las pacientes tratadas por cáncer de mama que son portadoras de mutación en BRCA1 tienen un riesgo acumulado de desarrollar cáncer de mama contralateral del 58% a los 50 años y de 65% hasta los 70 años de edad(120-122). Las mujeres portadoras del gen BRCA2 se estima que tienen riesgo en proporción similar pero con más bajo riesgo de cáncer de ovario.
Conforme se avanza en la investigación sobre la penetración de los genes BRCA1 y BRCA2 se estima que el riesgo de desarrollar cáncer de mama es algo menor que el descrito hasta ahora, así se desprende del estudio efectuado en la raza judía Ashkenazi(123) que estima un riesgo de cáncer de mama del 56% y de cáncer de ovario del 16,5%. Resulta de adicional interés en este estudio que no discrimina la localización de la mutación en el gen BRCA1 o BRCA2 se asocie con una variación en la gradación del riesgo para cáncer de ovario.
¿Existen diferencias clínicas entre el cáncer de mama hereditario y el esporádico?
Se analizan las diferencias clínicas y patológicas entre el cáncer hereditario y el esporádico, para ello Lynch y cols.(124) antes de disponer de los estudios genéticos de susceptibilidad al cáncer estudian 180 pacientes con síndrome familiar de cáncer de mama y ovario. En ochenta casos por las características clínicas peculiares es muy probable que hoy coincidan con las portadoras del gen BRCA1; pues bien en este grupo de pacientes se encuentran mayor proporción de tumores con aneuploidia y fracción de fase S elevada. Los tipos histológicos tubular y lobulillar son poco frecuentes asociados con mutaciones del gen BRCA1 y más frecuentes en mujeres portadoras de mutaciones del gen BRCA2. En estudios posteriores se demuestra aumento de frecuencia de tumores de tipo medular y medular atípico, con histología de pobremente diferenciados considerados de mal pronóstico por aumento del número de mitosis y pleomorfismo celular en las enfermas con mutación de la línea germinal BRCA1(125-128) y disminución de formación de túbulos e infiltración difusa de márgenes tumorales en el BRCA2(127). Paradójicamente, la probabilidad de supervivencia libre de enfermedad de los tumores relacionados con el gen BRCA1 presentan mejor supervivencia si se compara con los tumores esporádicos de similar grado. Esta contradicción se explica porque los tumores relacionados con los genes BRCA1 y BRCA2 responden con facilidad al tratamiento al tener un aumento de sensibilidad a radiaciones y algunos agentes quimioterápicos(129).
Los genes BRCA1 y BRCA2 son tumor supresores. Mecanismo de inactivación
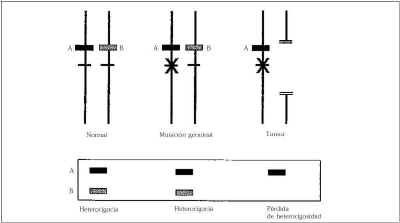
Todos los individuos nacen con dos copias de cada gen, sin embargo se necesita una explicación para el desarrollo de cáncer en un número grande de individuos que tienen como característica común tener sólo una mutación genética heredada en un gen tumor supresor, con una función normal en la otra copia. En 1971 Knudsen(130) tomando como ejemplo el gen del retinoblastoma emite la patogenia del cáncer humano y sugiere que el cáncer ocurre como resultado de dos procesos genéticos en la célula y como consecuencia se produce la inactivación de ambas copias del gen tumor supresor. En el caso de cáncer esporádico, no hereditario la probabilidad de que los dos acontecimientos se produzcan en la misma célula es pequeña. En los individuos con cáncer familiar se hereda una mutación que inactiva el gen tumor supresor en un alelo en todas las células del organismo debido a que se trata de una mutación de la línea germinal(131). Se requiere un proceso somático (no hereditario) para inactivar la copia restante con lo que se ocasiona el desarrollo del cáncer. Es decir, el resultado final es la pérdida o inactivación de ambos alelos, el primero por mutación de la línea germinal y el segundo por un proceso somático (Fig. 3). El cromosoma está representado por las dos líneas verticales, el gen tumor supresor (una copia por cada cromosoma materno y paterno) por las líneas cortas horizontales, el marcador genético de tamaño variable (A mayor que B) representado por un rectángulo está próximo al gen tumor supresor. La inactivación de una copia del gen tumor supresor se representa por una X en el gen por mutación hereditaria en la línea germinal, y la pérdida de la copia funcionante restante por la pérdida en el tumor de un segmento amplio del cromosoma que contiene el gen. En el interior del rectángulo debajo se expresan los marcadores genéticos identificados en un gel de electroforesis.

Figura 3.Pérdida de heterocigosidad. Representación esquemática del mecanismo de inactivación del gen tumor supresor que permite el desarrollo de una enfermedad enfermedad maligna.
En los síndromes de cáncer familiar el segundo proceso es posible detectarlo en muestras de tumor de mama por la pérdida de los marcadores en la región interesada del cromosoma, este fenómeno se denomina pérdida de heterocigosidad. Los trabajos recientes sobre pérdida de heterocigosidad en familias con cáncer de mama exhiben pérdida de heterocigosidad en el 20% de los tumores estudiados y localizados en los cromosomas: 8p, 16q, 17p, 17q y 19p. Aún más frecuentemente se demuestra la pérdida de heterocigosidad en el cromosoma 8p, en esta serie llega hasta el 33%(80).
En resumen, la susceptibilidad genética al cáncer de mama resulta de la inactivación de un alelo de BRCA1 por línea germinal y la subsiguiente pérdida somática del otro alelo en el tejido tumoral de la mama.
Espectro de las mutaciones del BRCA1 y BRCA2
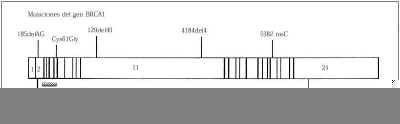
El gen BRCA1 es un gen grande lo que hace posible un enorme número de mutaciones, lo que dificulta la tecnología del diagnóstico. Desde la identificación del gen BRCA1 en 1994 (Fig. 4) se han detectado más de 300 mutaciones de las secuencias. Estas variantes se distribuyen a lo largo de todo el locus de la región del gen, y más del 50% de ellas solo se les ha identificado una sola vez. Las diferentes mutaciones se asocian con diferente riesgo de cáncer(59,100,132). Sin embargo ciertas mutaciones en el BRCA1 son más frecuentes en una población definida, así entre las mujeres judías Ashkenazi se identifica la mutación 185delAG con una frecuencia del 30%, y junto con las mutaciones 538insC y la 188del11(133) son las tres más frecuentemente observadas. En el estudio efectuado en 858 mujeres judías sanas se encuentra la mutación 185delAG en el 1% de la población(134), y la mutación 538insC en el 0,1% de esta población(135).

Figura 4. Representación esquemática del gen BRCA1. Mutaciones más frecuentes. Las líneas verticales representan los exones.
Existe controversia sobre si la localización de la mutación del gen BRCA1 en las consecuencias del individuo o de la familia. Dos estudios(59,132) sugieren que la mutación en la mitad 5'' del BRCA1 predispone a ambos cánceres de mama y ovario; mientras que las mutaciones más próximas en la porción 3'' del gen predominantemente se asocian sólo de cáncer de mama, pero no todos los estudios confirman estos hallazgos. También se especula que las mutaciones localizadas en la región terminal del gen BRCA1 se asocian con fenotipo más severo, definido por tumores de alto grado histológico(136).
Sorprendentemente todas las mutaciones identificadas hasta la fecha son mutaciones de la línea germinal, que afecta a todas las células del organismo, incluidas los óvulos y espermatozoides, y desde luego se transmiten a la descendencia en la siguiente generación. Las mutaciones adquiridas o somáticas del BRCA1 no se han descrito en los cánceres de mama humanos y sólo rara vez se describen en los tumores de ovario(137-139). Estos hallazgos permiten especular con la hipótesis de que el gen BRCA1 no sea un componente importante del desarrollo de tumores de mama y ovario, sino que la función del BRCA1 derive por otras vías y aparezca un cáncer de mama esporádico en vez de hereditario.
El gen BRCA2 es casi dos veces más grande que el BRCA1 lo que le hace más complejo para el diagnóstico de las mutaciones. Ya se han descrito más de 100 mutaciones del BRCA2(72,73,90,140-143) y algunas con el mismo espectro del BRCA1 simulando sus características. Como en las mutaciones del BRCA1, algunas mutaciones del gen BRCA2 son más frecuentes en algunas poblaciones, así entre las mujeres judías Ashkenazi la mutación 6174delT en BRCA2 se encuentra en el 1% de la población(100). La población de Islandia es pequeña y está bien definida porque se dispone de un registro de tumores junto con los antecedentes familiares; pues bien la mutación 999del5 del gen BRCA2 se encuentra en 16 mujeres de 21 familias con antecedentes de cáncer de mama(108). Los estudios de la población de Islandia muestra que la mutación se encuentra en el 7,7% de pacientes con cáncer de mama no seleccionadas y se estima una frecuencia del 0,6% en la población general(110,112). El riesgo estimado de cáncer de mama es del 37% a los 70 años de edad(144), riesgo que resulta más bajo que el del BRCA1.
OTROS SINDROMES GENÉTICOS EN RELACION CON EL RIESGO DE CANCER DE MAMA
Un número de síndromes familiares se relacionan con un riesgo elevado de cáncer de mama. Estos originan un pequeño número de pacientes con cáncer de mama y por lo general se les identifica por otras manifestaciones clínicas (Tabla 7).
Tabla 7 Rasgos seleccionados de un solo gen que predispone al cáncer de mama | ||
| Gen | Cromosoma | Manifestaciones clínicas |
| TP53 | 17p13.1 | Síndrome familiar de cáncer de Li-Fraumeni o SBLA (síndrome de sarcoma, mama, hueso, cerebro, pulmón, laringe, leucemia, corteza suprarrenal) |
| Cowden | 10q22-23 | Síndrome de hamartoma múltiple de Cowden (papilomas orales, pápulas faciales, queratosis palmar, neoplasia mamaria, tiroidea, de colon) |
| ATM | 11q22.3 | Ataxia telangiectasia (posible exceso de cáncer de mama en heterocigotos, portadores del gen) |
| Klinefelter | 47,XXY | El riesgo de cáncer de mama es similar al de la mujer normal |
| t(14;22) | 14-22 | Posible exceso de cáncer de mama en una translocación hereditaria relativamente frecuente |
Síndrome de Li-Fraumeni
El mejor conocido es el síndrome de Li-Fraumeni que se caracteriza por el desarrollo de gran variedad de cánceres que se presentan en niños y adultos, incluye cáncer de mama, sarcoma de tejidos blandos, tumor cerebral, osteosarcoma, leucemia, carcinoma de corteza suprarrenal y posiblemente otros tipos de tumores. Los miembros de esta familias desarrollan cáncer en edad temprana y con frecuencia presentan tumores múltiples. Las posibilidades de que cualquier miembro de la familia presente un cáncer es grande: 50% hacia los 30 años y más del 90% para los 70 años de edad(145). Este síndrome se relaciona con mutaciones de la línea germinal p53(146,147), se trata de un gen tumor supresor con las dos copias inactivadas en muchos tumores, lo que está de acuerdo con la hipótesis de Knudsen, es decir existe por vía hereditaria la mutación en una de las copias y se produce después la pérdida de la otra copia por otra causa distinta, y representa la frecuencia de mutaciones más alta en el cáncer de mama(148). Las mutaciones del p53 se relacionan con la pérdida de heterocigosidad en el cromosoma 17p13.1(149) y codifica una fosfoproteína nuclear de 53 Kda, cuyo papel principal es regular el ciclo celular y mantener la integridad del ADN(148). Si el ADN se daña, entonces aumentan los niveles de p53 y el ciclo celular se detiene en G1 para permitir la reparación.
Debido a la gran heterogeneidad del cáncer de mama se considera importante estudiar el espectro de mutaciones de p53 ya que estas mutaciones tienen especificidad histopatológica, por ejemplo no se encuentran mutaciones en el tumor mucinoso ni en el papilar infiltrante(150). También se describe relación entre mutaciones del p53 con el estadio avanzado del tumor(151), indicando que contribuye a la progresión del tumor y a una alta velocidad de proliferación celular.
Síndrome de Cowden
La enfermedad de Cowden se presenta con rareza, se caracteriza por múltiples lesiones mucocutáneas que incluyen triquilemomas faciales, queratosis de partes acras y papilomas orales, también se describen vitíligo y angiomas. La proliferación benigna en otros órganos, del tipo de hamartomas y tumores de localización variada como tiroides, mama, pólipos gastrointestinales, leiomiomas uterinos y lipomas(152,153). Las enfermedades benignas de la mama que se describen consisten en fibroadenoma, lesiones fibroquísticas, malformaciones de la areola y pezón y la hiperplasia epitelial(152,153).
En las publicaciones se observa una incidencia aumentada de cáncer de mama cuando se compara con la población general(152). El cáncer de mama se diagnostica en 10 de 21 pacientes femeninas, con cáncer bilateral en cuatro casos. El síndrome se considera que se hereda de forma autosómica dominante con variable expresividad(154).
Como muchas de las mujeres descritas en las publicaciones(153) viven pero con riesgo de desarrollar cáncer de mama el número de las afectadas se puede incrementar. Recientemente se identifica el gen de la enfermedad en el cromosoma 10q22-23(155), se trata de un gen tumor supresor en el que se describen ya varias mutaciones(156).
Síndrome de Peutz-Jeghers
El síndrome de Peutz-Jeghers es un trastorno con herencia autosómica dominante, que se caracteriza por la existencia de hamartomas gastrointestinales y pigmentación mucocutánea, los cuales no se consideran premalignos, se relaciona con el riesgo elevado de tumores gastrointestinales y no gastrointestinales(157,158), que incluyen al cáncer bilateral de mama(159).
Síndrome de Muir
El síndrome de Muir es una variante del síndrome tipo II de Lynch, se caracteriza por la asociación de múltiples tumores en la piel, tumores malignos del tracto digestivo inferior y aparato genitourinario(160-162). Muchas de las lesiones son habituales como carcinoma de células basales, acantomas, divertículos en colon que aparecen en la edad adulta como sucede en la población general. La herencia de este síndrome es autosómica dominante con alta penetración(161). Las mujeres con este síndrome presentan una tendencia aumentada de cáncer de mama, particularmente después de la menopausia(162). Se han descrito tres genes responsables del cáncer de colon(163-168). El síndrome de Muir resulta de la mutación de la línea germinal en los lugares MSH2 del cromosoma 2p(167,168) y MLH1 del cromosoma 3p21(166), son genes que afectan la capacidad de las células para reparar el daño al DNA.
Ataxia-telangiectasia
A diferencia de las alteraciones descritas con anterioridad, la ataxia telangiectasia es un síndrome autosómico recesivo. Los homocigotos se caracterizan por degeneración neurológica progresiva con ataxia cerebelosa, telangiectasias oculocutánea, hipersensibilidad a las radiaciones y un aumento de incidencia de enfermedades malignas. El gen de la ataxia-telangiectasia se relaciona con una sensibilidad anormal para radiación ionizante y los homocigotos tienen un riesgo aumentado 100 veces de cáncer. Por lo general, las pacientes mueren antes de los 20 años de edad de linfoma o leucemia(169). Los heterocigotos constituyen cerca del 1% de la población de Estados Unidos(170). Estos individuos tienen un mayor riesgo de cáncer en cualquier localización(171). Estudios epidemiológicos consideran en las portadoras femeninas del gen un riesgo mayor de cáncer de mama e incrementado en cinco veces. El diagnóstico de exposición ocupacional a la radiación ionizante se relaciona con un riesgo alto de cáncer de mama entre los portadores de ataxia telangiectasia(171). Recientemente se identifica el gen en el cromosoma 11q22-23(172), del que se describen unas seis mutaciones como más frecuentes, dos de ellas en familias del Japón son las más frecuentemente observadas(173). La ataxia-telangiectasia no contribuye de forma importante al riesgo familiar de cáncer de mama, pero si se puede considerar como un factor de riesgo de cáncer de mama(174).
Translocación constitucional de los cromosomas 11q;22q
En más de 100 familias se describe la translocación del brazo largo del cromosoma 11 con el brazo largo del cromosoma 22(175-177). La descripción de individuos con esta anomalía asciende al año 1980, pero hasta 1994 no se publica la existencia de enfermedades malignas, entonces es cuando se describen ocho casos de cáncer de mama en familias de Suecia con un total de 22 portadores(178).
PRUEBAS GENÉTICAS PARA SUSCEPTIBILIDAD DE CANCER DE MAMA
Los progresos recientes en la localización y secuencia de los genes que imparten susceptibilidad de cáncer de mama (BRCA1, BRCA2), así como la definición molecular de alteraciones hereditarias que se relacionan con el mayor riesgo de cáncer de mama han conducido a la identificación del umbral de susceptibilidad para este cáncer. Mediante pruebas de base molecular, la posibilidad de un individuo dado dentro de una familia de alto riesgo puede definirse con un alto grado de precisión, separando aquellos miembros de la familia que portan un factor de susceptibilidad y que tienen una probabilidad cercana al 100% de desarrollar cáncer de mama durante su vida, a diferencia de otros miembros de la familia que tienen un riesgo de cáncer de mama comparable con la población general.
El simple procedimiento de hacer una historia clínica indagando sobre los antecedentes familiares constituye una prueba genética. La denominada prueba genética predictiva indica análisis de DNA, RNA o proteínas de una persona sana para predecir el riesgo de presentar una enfermedad. En las pruebas predictivas para genes que predisponen al cáncer de mama, se utilizan dos estrategias ambas aprobadas por Institutional Review Boards.
Un método es el análisis de enlace genético, el llamado método de señales, que toma un grupo de marcadores genéticos y los utiliza como señales individualizadas. Se escogen marcadores de manera que su modelo sea diferente entre las personas. El propósito es buscar el cromosoma específico que porta el gen predisponente al cáncer de mama en una familia, para verificar que el modelo de esta neoplasia está relacionado con el cromosoma específico (cromosoma 17 para BRCA1). Si se observa un tipo definido en la generación previa y en las personas afectadas, entonces la mujer sana más joven de la misma familia, se estudia para el mismo modelo específico. Este método precisa una muestra del DNA de dos personas afectadas y dos no afectadas, requerimiento que a menudo necesita autorización para utilizar los bloques de tejido patológico de archivo y extraer DNA del tejido fijado de una mujer que tal vez murió de cáncer de mama. Con esta técnica es posible hacer predicciones con gran confianza en cuanto a quien es portador del gen predisponente.
El segundo método es el de las pruebas intragénicas, que responden a la pregunta ¿cambió la secuencia de DNA en un punto? Se requiere sólo un análisis y si es positivo para un cambio o mutación, responde la pregunta. Un ejemplo es la mutación más frecuentemente observada en judíos Ashkenazi(134) del gen BRCA1 entre los nucleótidos de las posiciones 185 y 186 hay una delección de dos de ellos, adenosina y guanosina, que se abrevia con la denominación 185delAG. Si se encuentra la mutación en una mujer judía con cáncer de mama, es probable que el gen sea el principal determinante de su cáncer y es posible estudiarlo fácilmente en otras mujeres de la familia. Si el resultado es negativo, la mujer puede tener una mutación en otra parte de BRCA1, en otro gen o ninguna predisposición genética conocida y entonces la respuesta es incierta. Por tanto hoy es técnicamente imposible hacer a una mujer la prueba para el gen del cáncer de mama. No se trata de una sola prueba; ni siquiera hay un solo gen predisponente.
Cualquiera de los dos métodos utilizados implica a más de un miembro de la familia; es decir, el riesgo de cáncer hereditario es un asunto familiar, haya o no parientes que la admitan. Además de los problemas de laboratorio, los resultados suelen no ser todo o nada, con un riesgo de 100% o 0%. Más bien los resultados suelen modificar probabilidades, un concepto difícil de asimilar por las pacientes y los clínicos. Por ejemplo, se sabe que la presencia del 185 del AG está vinculado con un riesgo de 85% de presentar cáncer de mama, pero la ausencia de mutación en la hermana no significa que no tendrá cáncer de mama, sino más bien que su peligro se iguala al riesgo de la población.
Debido a estas complejidades, el consenso actual es que la valoración del riesgo, el asesoramiento y la toma de decisiones inclusive para considerar las pruebas genéticas, son temas para clínicas especializadas. A pesar de este consenso, la American Society of Clinical Oncology ha dado a los oncólogos en ejercicio la luz verde para proceder con las pruebas(179).
RIESGO MODERADO DE CANCER DE MAMA
Para la mayoría de las mujeres con una historia familiar de cáncer de mama que caen dentro de la categoría de riesgo moderado, las pruebas genéticas para susceptibilidad de cáncer de mama no son aconsejables.
En el asesoramiento del riesgo, cuando no se reconoce una predisposición mendeliana ni es posible hacer estudios genéticos, el cálculo empírico del riesgo es posible realizarlo por uno de los dos modelos considerados como estándar. El modelo Claus o modelo CASH tiene en cuenta los antecedentes familiares de cáncer de mama en uno o dos miembros de la familia de primer y segundo grado, como se describe en la tabla 2. En el modelo Gail(60) se selecciona un número de los grupos A, B y C de la tabla 8 para incorporar cuatro factores de riesgo importantes: edad de la menarquia, edad del primer parto a término, número de biopsias de mama y número de parientes de primer grado con cáncer de mama. Estos tres números se multiplican para obtener el riesgo relativo conjunto, que se multiplica entonces por 0,93, si en ninguna biopsia se demuestra hiperplasia atípica o por 1,82 cuando si existe.
Tabla 8 Peligro relativo de factores de riesgo seleccionados(55)* | ||
| Factor de riesgo | N.º de pacientes de primer grado con cáncer de mama | Riesgo relativo |
| Grupo A. Edad menarquia (años) | ||
| > 14 | 1,00 | |
| 12 a 13 | 1,10 | |
| < 12 | 1,21 | |
| Grupo B. Número de biopsias mamarias. | ||
| Edad < 50 años en el momento del asesoramiento | ||
| 0 | 1,00 | |
| 1 | 1,70 | |
| > 2 | 2,88 | |
| Edad >50 años en el momento del asesoramiento | ||
| 0 | 1,00 | |
| 1 | 1,27 | |
| > 2 | 1,62 | |
| Grupo C. Edad en el primer parto a término (años) | ||
| < 20 | 0 | 1,00 |
| 1 | 2,61 | |
| > 2 | 6,80 | |
| 20 a 24 | 0 | 1,24 |
| 1 | 2,68 | |
| > 2 | 5,78 | |
| 25 a 29 o nulíparas | 0 | 1,55 |
| 1 | 2,76 | |
| > 2 | 4,91 | |
| > 30 | 0 | 1,93 |
| 1 | 2,83 | |
| > 2 | 4,17 | |
Este concepto matemático de riesgo es difícil de comprender y lo mejor es traducirlo a una probabilidad absoluta. Las matemáticas que se requieren son complejas ya que se utiliza una ecuación de peligros múltiples para resumir el riesgo anual de no tener cáncer de mama. Para simplificar el cálculo, se preparan gráficas que traducen el riesgo relativo a uno estimado de la probabilidad en un período de 30 años.
En el modelo Gail no se utiliza la historia familiar ni la edad de diagnóstico en la valoración del riesgo; sin embargo toma en consideración otros factores de riesgo epidemiológicos conocidos. Los propósitos específicos de los modelos de Gail y Claus son diferentes. El primero supone una población de mujeres blancas que se estudian cada año para cáncer de mama y señala ciertos factores de riesgo y el número de parientes de primer grado con cáncer mamario para proyectar un riesgo individualizado de la enfermedad. El modelo de Claus utiliza un modo supuesto de herencia para susceptibilidad a la neoplasia maligna de mama y calcula el riesgo de quienes tienen antecedente familiar. Para asesorar mujeres a las que se hace análisis de riesgo de cáncer de mama lo mejor es tal vez calcular los riesgos a partir de ambos modelos y ofrecer a la paciente una variación de cifras de riesgo en contraposición a un solo riesgo numérico.
ESTRATEGIAS PARA REDUCIR EL RIESGO DE CANCER DE MAMA
La mayoría de los factores que influyen el riesgo femenino de cáncer de mama como herencia, edad, edad de inicio de la menarquia y de la menopausia está lejos de cualquier control. Otros factores como la elección de tener un embarazo antes de los 20 años de edad o el incremento en la paridad no se modifican con facilidad por razones obvias. Las modificaciones en el estilo de vida (dieta adecuada pobre en grasas animales, ejercicio y reducción del consumo de alcohol) no se relacionan de manera convincente con una disminución del riesgo de cáncer de mama, pero se recomiendan como medidas consistentes con una buena salud general. De cualquier manera el clínico debe estar preparado para aconsejar a las mujeres y darles a entender como la historia familiar de cáncer de mama impacta en su riesgo individual. Esto es de vital importancia para dar libertad a las mujeres que tomen decisiones informadas en relación con riesgos adicionales y beneficios potenciales que puedan incurrir a partir del uso de estrógenos exógenos (anticonceptivos orales, inducción de la ovulación, estrógenos sustitutivos de la menopausia). Para las mujeres con un riesgo moderado de cáncer de mama, la detección rutinaria de acuerdo con las guías recomendadas es la estrategia más importante para asegurar la detección precoz, ya que hasta la fecha los esfuerzos para diseñar programas de detección con base en factores de riesgo no son productivos, porque la estimación de la efectividad en las mujeres diana con base en un grupo de factores de riesgo clave tales como historia familiar, edad de la menarquia y paridad ha demostrado que estos programas fallan en la mayoría de los casos. Desde el punto de vista de salud pública, todas las mujeres en una categoría apropiada de edad deben explorarse de acuerdo con las normas recomendadas.
DISCUSION DEL RIESGO
El riesgo de padecer cáncer de mama se acentúa por la existencia de antecedentes familiares afectados, resultando la paciente o la familia de riesgo moderado o alto de acuerdo con los conceptos que venimos diciendo, o por los resultados histológicos de una biopsia de mama en la propia paciente considerada como hiperplasia atípica o carcinoma lobulillar in situ. Cuando a una mujer sana dentro de una familia de alto riesgo se le detecta una mutación del gen BRCA1 se le considera como portadora del gen de susceptibilidad al cáncer, pero el resultado es solo de título informativo del riesgo probable. Al contrario, cuando el test es negativo el resultado produce menos información porque indica la posibilidad de que exista otro gen diferente como responsable o no existe predisposición hereditaria y por tanto la mujer no tiene riesgo adicional.
Algunas mujeres cuando se les solicita permiso para realizar pruebas de susceptibilidad al cáncer prefieren no realizar el test de diagnóstico de la mutación(180,181). El conocimiento del riesgo produce un cambio emocional en la paciente, que se describe ampliamente en una revisión los aspectos psicológicos(182) e incluso se describen reacciones psicológicas adversas después de tal información como son ansiedad, depresión, culpabilidad y reducción de la propia estima.
OPCIONES DE TRATAMIENTO
Una vez conocidos el riesgo alto de padecer cáncer de mama el tratamiento implica la vigilancia, quimioprevención y mastectomía profiláctica, aunque existen pocos datos de que estas opciones reduzcan la mortalidad en enfermas de alto riesgo.
Vigilancia
La vigilancia atenta de la mujer es muchas veces la opción elegida. Aunque no existen datos de ensayos prospectivos sobre la eficacia de la vigilancia en pacientes de alto riesgo se adopta esta vía por extrapolación del conocimiento que el esfuerzo de una detección precoz reduce la mortalidad en mujeres de más de 50 años. La propuesta es de Lynch(183), basada en la experiencia propia de vigilancia de familias de alto riesgo, para ello recomienda empezar las exploraciones muy pronto, unos diez años antes del diagnóstico del familiar más joven afectado. Se recomienda la mamografía anual y exploración clínica cada seis meses y en algún caso muy especial se recomienda la mamografía cada seis meses.
Quimioprevención
Se dispone en la actualidad de resultados de tres estudios aleatorizados(184-186) en mujeres con riesgo de cáncer de mama empleando la toma de 20 mg/día de tamoxifeno durante cinco años (Tabla 9). El estudio americano(184) selecciona las pacientes para entrar en el protocolo del National Surgical Adjuvant Breast and Bowel Project (NSABP) (P-1) mediante el método de Gail, tras un seguimiento de 55 meses se interrumpe el ensayo unos catorce meses antes de completar el tiempo previsto porque se considera que el efecto protector del tamoxifeno está suficientemente probado no necesitándose mayor seguimiento. El número de cánceres de mama aparecido en las mujeres tratadas es prácticamente la mitad de los acontecidos en el grupo control lo que representa un 49% de reducción del riesgo, que llega al 69% para los cánceres con receptores de estrógeno positivos. El estudio inglés(185) y el italiano(186) concluyen que el tamoxifeno no protege del cáncer de mama, pero en ambos la selección de pacientes es diferente, se incluyen mujeres más jóvenes y pacientes en tratamiento con estrógenos sustitutivos de la menopausia, además en el ensayo inglés se admiten mujeres de riesgo por los antecedentes de familiares afectados y en el italiano la condición precisa de mujeres histerectomizadas. Quedan sin contestar algunas preguntas como es la duración del efecto protector del tamoxifeno en la reducción del riesgo, el efecto del tamoxifeno sobre mujeres portadoras de los genes BRCA1 y BRCA2 y el efecto sobre el riesgo de muerte por cáncer.
Tabla 9 Quimioprevención del cáncer de mama con tamoxifeno | |||||
| Ensayo | N.º de mujeres | Seguimiento (mujeres/año) | Cáncer de mama en tamoxifeno/control | Cáncer de mama/1.000 mujeres/años | |
| Tamoxifeno | Control | ||||
| NSABP 184 | 13.388 | 46.858 | 116/213 | 3,6 | 6,6 |
| Inglés 185 | 2.471 | 12.355 | 34/36 | 4,7 | 5,0 |
| Italiano 186 | 5.408 | 20.731 | 19/22 | 2,1 | 2,3 |
Mastectomía profiláctica
La mastectomía profiláctica en pacientes de alto riesgo es de una eficacia desconocida porque no se dispone de ensayos prospectivos aleatorizados que idealmente deberían comparar dos técnicas, una la mastectomía, la otra vigilancia o quimioprevención con tamoxifeno y con seguimiento largo de 10 y hasta 20 años.
Los datos disponibles se basan en estudios retrospectivos con indicaciones médicas que no siempre incluyen pacientes de alto riesgo, pero se dispone de seguimiento largo de hasta 14 y 17 años con persistencia del beneficio. Los cánceres que se describen aparecen como recidivas, lo hacen en la pared torácica y en axila(187,188), y no se describen metástasis a distancia.
Con la técnica de mastectomía subcutánea se describe persistencia de tejido mamario como se demuestra en una serie de necropsias(189), el tejido mamario residual se localiza fundamentalmente en la región periareolar y en márgenes periféricos. Por este motivo es recomendable la mastectomía total con prótesis subpectoral de suero fisiológico o por injertos de colgajos próximos. La tabla 10 resume la efectividad de la mastectomía profiláctica, estimada en el 99% de reducción de la incidencia de cáncer de mama. En la revisión reciente(193) se describe la reducción del riesgo en pacientes de riesgo moderado del 89,5% y la aparición de tres cánceres entre 214 pacientes de alto riesgo con incidencia de 1,4%.
Tabla 10 Efectividad de la mastectomía profiláctica | |||||
| Autor | Número total | Número de alto riesgo | Tipo de mastectomía | Seguimiento (años) | Número de cánceres |
| Ziegler 188 | 285 | 16 | Subcutánea | ns | 3 |
| Jarret. 190 | 100 | ns | Total | > 5 | |
| Pennisi 191 | 1.500 | 735 | Subcutánea | 9 | 6 |
| Woods 192 | 1.400 | ns | Subcutánea | 17 | 3 |
| Hartmann 193 | 950 | 213 | ns | 14 | 7 |
| ns = no se conoce. | |||||
Por otra parte se describe el grado de satisfacción de la mastectomía en pacientes de alto riesgo, porque se reduce la ansiedad(194), como se demuestra en el seguimiento de 14 mujeres a las que se practica mastectomía profiláctica, son especialmente seleccionadas por tener alto riesgo, mayor número de biopsias de mama y el más alto índice de riesgo cuantificado, entre las 164 pacientes seguidas por tener al menos un familiar afectado.
RECOMENDACIONES PARA LA MUJER CON SUSCEPTIBILIDAD HEREDITARIA AL CANCER DE MAMA
En el momento actual existe controversia e incertidumbre sobre el tratamiento de la mujer sana portadora de susceptibilidad hereditaria al cáncer de mama. No se conoce que la vigilancia y seguimiento de estas mujeres reduzca la mortalidad de los cánceres relacionados en las mujeres de alto riesgo. Además se entiende que la vigilancia clínica y la mamografía no son capaces de detectar lesiones premalignas, ni siquiera les ofrece protección. Las propias mujeres nos preguntan acerca del valor profiláctico de la mastectomía en ausencia de cualquier otra opción preventiva. Desde el punto de vista teórico parece lógico que una vez eliminado el tejido en riesgo se reduce la probabilidad del desarrollo de cáncer, pero hay pocos datos en que se demuestre la eficacia de la mastectomía en este sentido. Además existe la evidencia de que la técnica quirúrgica no extirpa todo el tejido mamario ni siquiera con la mastectomía total, es posible que la mutación de la línea germinal esté presente en el tejido mamario residual y la paciente quede en riesgo de padecer cáncer de mama. De manera similar sucede con la ovariectomía que la técnica no garantiza la protección absoluta frente al cáncer de ovario, porque es posible la generación de tumores a partir de la superficie peritoneal, con el riesgo estimado entre el 2 y 25%(195,196). En este sentido existe la especulación sobre si los tumores de la superficie peritoneal son en realidad producto de metástasis de cáncer de ovario no diagnosticado o de una lesión primaria. Esta última explicación se ajusta a la realidad anatómica del origen del epitelio que recubre el ovario, que es la simple capa celular del peritoneo que se refleja en el ovario y lo recubre. Se precisa de estudios prospectivos sobre el papel profiláctico de la mastectomía y de la ovariectomía en las mujeres portadoras de los genes BRCA1 y BRCA2. Estas dudas dificultan el consejo a la paciente portadora sobre el beneficio potencial de las técnicas quirúrgicas para no ofrecer un falso sentido de seguridad.
En la mujer que tiene un diagnóstico de mutación del gen BRCA1 el Colegio Americano de Obstetricia y Ginecología recomienda la ovariectomía bilateral cuando se completa la descendencia o en el momento de la menopausia, porque no existe un programa de diagnóstico precoz de cáncer de ovario.
BIBLIOGRAFIA
1. Kelsey JL, Gammon M. The epidemiology of breast cancer. Cancer 1991;41:146-65.
2. Kelsey JL. The epidemiology of breast cancer. Epidemiol Rev 1993;15:256-63.
3. Bernstein L, Henderson BE, Hanisch R y cols. Physical exercise and reduced risk of breast cancer in young woman. J Natl Cancer Inst 1994;86:1403-8.
4. Anderson DE. Some characteristics of familial breast cancer. Cancer 1971;28:1500-4.
5. Schwartz AG, King MC, Belle SH y cols. Risk of breast cancer to relatives of young breast cancer patients. J Natl Cancer Inst 1985;75:665-8.
6. Sattin RW, Rubin GL, Webster LA y cols. Family history and the risk of breast cancer. JAMA 1985;253:1908-13.
7. Byrne C, Brinton LA, Haile RW y cols. Heterogeneity of the effect of family history on breast cancer risk. Epidemiology 1991;2:276-84.
8. Ries LAG, Miller BA, Hankey BF y cols. Eds SEER. Cancer statistics review 1973-1991: tables and graphs. NIH publ no 94-2789. Bethesda. USDHHS. National Cancer Institute, 1994.
9. Bernstein JL, Thompson WD, Risch N, Holford TR. The genetic epidemiology of second primary breast cancer. Am J Epidemiol 1992;136:937-48.
10. Toniolo PG. A prospective study of endogenous estrogens and breast cancer in postmenopausal women. J Natl Cancer Inst 1995;87:190-7.
11. Henderson BE, Pike MC, Casagrande JT. Breast cancer and the oestrogen window hypotesis (letter). Lancet 1981;2:363-264.
12. Henderson BE, Ross RK, Judd HL y cols. Do regular ovulatory cycles increase breast cancer risk? Cancer 1985;56:1206-8.
13. De Waard F. Diet and breast cancer. En: Benito E, Giacosa A, Hill MJ (eds). Public education on diet and cancer. Kluwer academic publishers, 1991.
14. Pike MC, Berstein L, Spicer DV. Exogenous hormones in breast cancer risk. En: Mederhuber JE (ed). Current therapy in Oncology. MO Decker. St Louis 1993;292-303.
15. Brinton LA, Schairer CS, Hoover RN y cols. Menstrual factors and risk of breast cancer. Cancer Invest 1988;6:245-54.
16. Vatten L, Kvinnslands S. Pregnancy-related factors and risk of breast cancer in a prospective study of 29.981 norwegian women. Eur J Cancer 1992;28A:1148-53.
17. MacMahon B, Cole P, Lin TM y cols. Age at first birth and breast cancer risk. Bull World Hlth Org 1970;43:209-21.
18. Newcomb PA, Storer BE. Lactation and lower incidence of premenopause breast cancer. N Engl J Med 1994;330:81-7.
19. Thomas DB, Noonan EA. Breast cancer and extended lactation. Int J Epidemiol 1993;22:619-26.
20. Yuan JM, Yu MC, Ross RK y cols. Risk factors for breast cancer in chinese women in Shanghai. Cancer Res 1988;48:1949-53.
21. Melbye M. Induced abortion and risk of breast cancer. N Engl J Med 1997;336:81-5.
22. Trichopoulos D, MacMahon B, Cole P. Menopause and breast cancer risk. J Natl Cancer Inst 1972;48:605-13.
23. Goodman MJ. Breast cancer in multi-ethnic populations: The Hawai perspective. Breast Cancer Res Treat 1991;18,supl 1:55-9.
24. Ziegler RG, Hoover RN, Pike MC y cols. Migration patterns and breast cancer risk in Asian-American women. J Natl Cancer Inst 1993;85:1819-27.
25. Krieger N. Social class and the black-white cross-over in the age-specific incidence of breast cancer: A study linking census derived data to population-based registry records. Am J Epidemiol 1990;131:804-14.
26. De Waard F, Cornelis JP, Aoki K, Yoshida M. Breast cancer incidence according to weight and height in two cities of the Netherlands and Aichi prefecture, Japan. Cancer 1977;40:1269-75.
27. Ingram D, Sanders K, Kolybata M, López D. Case-control study of phytoestrogens and breast cancer. Lancet 1997;350:990-4.
28. Vatten LJ, Kvinnsland S. Body height and risk of breast cancer: A prospective study of 23.831 Norwegian women. Br J Cancer 1990;61:881-5.
29. Vatten LJ, Kvinnsland S. Prospective study of height, body mass index and risk of breast cancer. Acta Oncol 1992;31:195-200.
30. Longnecker MP, Berlin JA, Orza MJ y cols. A meta-analysis of alcohol consumption in relation to breast cancer risk. JAMA 1988;260:652-6.
31. Bernstein L, Henderson BE, Hanisch R y cols. Physical exercise and reduced risk of breast cancer in young women. J Natl Cancer Inst 1994;861403-8.
32. Thune Y, Brenn T, Lund E, Gaard M. Physical activity and the risk of breast cancer. N Engl J Med 1997;336:1269-75.
33. Dorgan JF, Brown C, Barrett M, Y cols. Physical activity and risk of breast cancer in the Framingham heart study. Am J Epidemiol 1994;139:662-9.
34. Dupont WD, Page DI. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985;312:146-51.
35. London SJ, Connolly JL, Schnitt SJ, Colditz GA. A prospective study of benign breast disease and the risk of breast cancer. JAMA 1992;267:941-4.
36. González Merlo J. Tratamiento hormonal sustitutivo y cáncer de mama. En: Libro del año en Obstetricia y Ginecología. Saned 1996:49-58.
37. Comino R, Fernández JJ, Lubian D. Tratamiento hormonal sustitutivo y cáncer de mama. Prog Obst Gin 1997;5:303-30.
38. Delgado M, Sillero-Arenas M, Sánchez R. Terapia hormonal sustitutiva en la menopausia y riesgo de cáncer de mama. Un meta-análisis. Prog Obst Gin 1994;5:415-21.
39. Brinton LA, Gammond MD, Malone KE y cols. Modification of oral contraceptives relationships on breast cancer risk by selected factor among younger women. Contraception 1997;55:197-203.
40. Breast cancer and hormonal contraceptives: Collaborative reanalysis of individual data on 53.297 women with breast cancer and 100.239 women without breast cancer from 54 epidemiological studies. Collaborative group on hormonal factors in breast cancer. Lancet 1996;347:1713-27.
41. White E, Malone KE, Weis NS. Breast cancer among young US women in relation to oral contraceptive use. J Natl Cancer Inst 1994;86:505-14.
42. Van-os Wa, Edelman DA, Rhemrev PE, Grant S. Oral contraceptives and breast cancer risk. Adv Contracept 1997;13:63-9.
43. Rushton L, Jones DR. Oral contraceptive and breast cancer risk. Br J Obstet Gynaecol 1992;99:239-46.
44. John EM, Kelsey J. Radiation and other environmental exposures and breast cancer. Epidemiol Rev 1993;15:157-62.
45. Hancock SL, Tucker MA, Hoppe RT. Breast cancer after treatment of Hodgkin''s disease. J Natl Cancer Inst 1993;85:25-31.
46. Anderson DE. Genetic study of breast cancer: Identification of a high risk group. Cancer 1974;34:1090-7.
47. Anderson DE, Badzioch MD. Risk of familial breast cancer. Cancer 1985;56:383-7.
48. Sanjosé S, Viladiu P, Cordon F y cols. Cáncer de mama y herencia: Resultados de un estudio poblacional de casos y controles en Girona. Med Clin (Barc) 1998;110:370-2.
49. Goldhirsch A, Glick JH, Gelber RD, Senn HJ. Meeting highlights: International consensus panel on the treatment of primary breast cancer. J Natl Cancer Inst 1998;90:1601-8.
50. Hoskins KF, Stopfer JE, Calzone KA y cols. Assessment and counseling for women with a family history of breast cancer: A guide for clinicians. JAMA 1995;273:577-85.
51. Claus EB, Risch N, Thompson WD. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet 1991;48:232.
52. Mulvihill JJ, Safyer AW, Bening JK. Prevention in familial breast cancer: Counseling and prophylactic mastectomy. Prev Med 1982;11:500-11.
53. Ottman R, Pike MC, King MC y cols. Practical guide for estimating risk for familial breast cancer. Lancet 1983;2:556-8.
54. Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early onset breast cancer: Implications for risk prediction. Cancer 1994;73:643-51.
55. Gail MH, Brinton LA, Byar DP y cols. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989;81: 1879-86.
56. Benichou J. A computer programe for estimating individualized probabilities of breast cancer. Comput Biomed Res 1993;26:373-82.
57. Gail MH, Benichou J. Epidemiology and biostatistics program of the National Cancer Institute. J Natl Cancer Inst 1994;86:573-5.
58. Baker LH. Breast cancer detection demonstration project: Five-year summary report. Cancer J Clin 1982;32:194-225.
59. Benichou J, Gail MH, Mulvihill JJ. Graphs to estimate an individualized risk of breast cancer. J Clin Oncol 1996;14:103-10.
60. Spiegelman D, Colditz GA, Hunter D y cols. Validation of the Gail et al model for predicting individualized breast cancer risk. J Natl Cancer Inst 1994;86:600-7.
61. Bondy ML, Lustbader DE, Halabi S y cols. Validation of a breast cancer risk assessment model in women with a positive family history. J Natl Cancer Inst 1994;86:620-5.
62. Smigel K. Breast cancer Prevention trial takes off. J Natl Cancer Inst 1992;84:669-70.
63. Slattery ML, Kerber RA. A comprehensive evaluation of family history and breast cancer risk: The Utah population data base. JAMA 1993;270:1563-8.
64. Narod SA, Goldgar D, Cannon-Albright L y cols. Risk modifiers in carriers of BRCA1 mutations. Int J Cancer 1995;65:394-8.
65. Lynch HT, Harris RE, Guirgis HA y cols. Familial association of breast/ovarian cancer. Cancer 1978;41:1543-9.
66. Peto J, Easton DE, Mattews EE y cols. Cancer mortality in relatives of woman with breast cancer: The OPCS study. Int J Cancer 1996;65:275-83.
67. Thompson WD, Schildkrant JM. Family history of gynecologic cancers: Relationships to the incidence of breast cancer prior to age 55. Int J Epidemiol 1991;20:595-602.
68. Burke W, Daly M, Garber J y cols. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2. Cancer Genetic Studies Consortium. JAMA 1997;277:997-1003.
69. Hall JM, Lee MK, Newman B y cols. Linkage analysis of aerly onset breast cancer to chromosome 17q21. Science 1990;250:1684-9.
70. Narod SA, Feunteun J, Lynch HT y cols. Familial breast-ovarian cancer locus on chromosome 17q12-23. Lancet 1991;338:82-3.
71. Miki Y, Swensen J, Shattuck-Eidens D y cols. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66-71.
72. Wooster R, Bignell G, Lancaster J y cols. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995;378:789-92.
73. Tavtigian SV, Simard J, Rommens J y cols. The complete BRCA2 gene and mutations in 13q linked kindreds. Nat Genet 1996; 12:333-7.
74. Serova OM, Mazoyer S, Puget N y cols. Mutations in BRCA1 and BRCA2 in breast cancer families. Are there more breast cancer susceptibility genes? Am J Hum Genet 1997;60:486-95.
75. Ford D, Easton DE. The genetics of breast and ovarian cancer. Br J Cancer 1995;72:805-12.
76. Wooster R, Neuhausen SL, Mangion J y cols. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994;265:2088-92.
77. Ramus SJ, Kote-Jarai Z, Friedman LS y cols. Analysis of BRCA1 and BRCA2 mutations in Hungarian families with breast or breast-ovarian cancer. Am J Hum Genet 1997;60:1242-6.
78. Hakansson S, Johannsson O, Johansson U y cols. Moderate frequency of BRCA1 and BRCA2 germ-line mutation in Scandinavian familial breast cancer. Am J Hum Genet 1997;60:1068-78.
79. Schubert EL, Lee MK, Mefford HC y cols. BRCA2 in American families with four or more cases of breast or ovarian cancer: Recurrent and novel mutations, variable expression, penetrance and the possibility of families not attributable to BRCA1 or BRCA2. Am J Hum Genet 1997;60:1031-40.
80. Lindblom A, Skoog L, Rotstein S y cols. Loss of heterozygosity in familial breast carcinoma. Cancer Res 1993;53:4356-61.
81. Gayther SA, Harrington P, Russell P y cols. Frequently occurring germline mutations of the BRCA1 gene in ovarian cancer families from Russia. Am J Hum Genet 1997;60:1239-42.
82. Levy-Lahad E, Catane R, Eisenberg S y cols. Recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: Frequency and diferencial penetrance in ovarian cancer and in breast-ovarian cancer families. Am J Hum Genet 1997;60:1059-67.
83. Simard J, Tonin P, Durocher F y cols. Common origins of BRCA1 mutations in Canadian breast and ovarian cancer families. Nat Genet 1994;8:392-8.
84. Phelan C, Lancaster J, Tonin P y cols. Mutation analysis of the BRCA2 gene in 49 site-specific breast cancer families. Nat Genet 1996;13:120-2.
85. Castilla LH, Couch FJ, Erdos MR y cols. Mutations in the BRCA1 gene in families with aerly-onset breast and ovarian cancer. Nat Genet 1994;8:387-91.
86. Friedman LS, Ostermeyer EA, Szabo CI y cols. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat Genet 1994;8:399-404.
87. Friedman LS, Szabo CI, Ostermeyer EA y cols. Novel inherited mutations and variable expressivity of BRCA1 alleles, including the founder mutation 185delAG in Ashkenazi Jewish families. Am J Hum Genet 1995;57:1284-97.
88. Streuwing JP, Brody LC, Erdos MR y cols. Detection of eight BRCA1 mutations in 10 breast/ovarian cancer families, including 1 family with male breast cancer. Am J Hum Genet 1995;57:1-7.
89. Arena JF, Smith S, Plewinska M y cols. BRCA1 mutations in African-American women. Am J Hum Genet 1996;59:A34.
90. Couch F, Farid L, DeShano M y cols. BRCA2 germline mutations in male breast cancer cases and breast cancer families. Nat Genet 1996;13:123-5.
91. Serova OM, Montagna M, Torchard D y cols. A high incidence of BRCA1 mutations in 20 breast-ovarian cancer families. Am J Hum Genet 1996;58:42-51.
92. Gao Q, Neuhausen S, Cummings S y cols. Recurrent germline BRCA1 mutations in extended African American families with early-onset breast cancer. Am J Hum Genet 1997;60:1233-6.
93. Caligo MA, Ghimenti C, Cipollini G y cols. BRCA1 germline mutational spectrum in Italian families from Tuscany: A high frequency of novel mutations. Oncogene 1996;13:1483-8.
94. Montagna M, Santacatterina M, Corneo B y cols. Identification of seven new BRCA1 germline mutations in Italian breast and breast-ovarian cancer families. Cancer Res 1996;56:5466-9.
95. De Benedetti V, Radice P, Mondini P y cols. Screening for mutations in exon 11 of the BRCA1 gene in 70 Italian breast and ovarian cancer patients by protein truncation test. Oncogene 1996;13:1353-7.
96. Serova-Sinilnikova OM, Boutrand L, Stoppa-Lyonnet D y cols. BRCA2 mutations in hereditary breast and ovarian cancer in France. Am J Hum Genet 1997;60:1236-9.
97. Stoppa-Lyonnet D, Laurent-Puig P, Essioux L y cols. BRCA1 sequence variations in 160 individuals referred to a breast-ovarian family cancer clinic: Am J Hum Genet 1997;60:1021-30.
98. Johannsson O, Ostermeyer EA, Hakansson S y cols. Founding BRCA1 mutations in hereditary breast and ovarian cancer in southern Sweden. Am J Hum Genet 1996;58:441-50.
99. Gayther S, Harrington P, Russell P y cols. Rapid detection of regionally clustered germline BRCA1 mutations by multiplex heteroduplex analysis. Am J Hum Genet 1996;58:451-6.
100. Gayther S, Mangion J, Russell P y cols. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet 1997;15:103-5.
101. Xu CF, Chambers J, Nicolai H y cols. Mutations and alternative splicing of the BRCA1 gene in UK breast-ovarian cancer families. Genes Chromosom Cancer 1997;18:102-10.
102. Jandrig B, Grade K, Seitz S y cols. BRCA1 mutations in German breast-cancer families. Int J Cancer 1996;68:188-92.
103. Hamann U, Brauch H, Garvin AM y cols. German family study on hereditary breast and/or ovarian cancer: Germline mutation analysis of the BRCA1 gene. Genes Chromosom Cancer 1997;18:126-32.
104. Hogervorst FBL, Cornelis RS, Bout M y cols. Rapid detection of BRCA1 mutations by the protein truncation test. Nat Genet 1995;10:208-12.
105. Peelen T, Van Vliet M, Petrij-Bosch A y cols. A high proportion of novel mutations in BRCA1 with strong founder effects among Dutch and Belgian hereditary breast and ovarian cancer families. Am J Hum Genet 1997;60:1041-9.
106. Andersen TI, Borresen AL, Moller P. A common BRCA1 mutation in Norwegian breast and ovarian cancer families? Am J Hum Genet 1996;59:486-7.
107. Inoue R, Fukutomi T, Ushijima T y cols. Germline mutation of BRCA1 in Japanese breast cancer. Cancer Res 1995;55:3521-4.
108. Thorlacius S, Olafsdottir G, Tryggvadottir L y cols. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat Genet 1996;13:117-9.
109. Vehmanen P, Friedman LS, Eerola H y cols. A low proportion of BRCA2 founder mutations in Finnish breast cancer families. Am J Hum Genet 1997;60:1050-8.
110. Thorlacius S, Sigurdsson S, Bjarnadottir H y cols. Study of a single BRCA2 mutation with a high carrier frequency in a small population. Am J Hum Genet 1997;60:1079-84.
111. Friedman LS, Gayther SA, Kurosaki T y cols. Mutation analysis of BRCA1 and BRCA2 in a male breast cancer population. Am J Hum Genet 1997;60:313-9.
112. Shattuck-Eidens D, Oliphant A, McClure M y cols. BRCA1 sequence analysis in women at high risk for susceptibility mutations. JAMA 1997;278:1242-50.
113. Couch FJ, DeShano M, Blackwood MA y cols. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 1997;336:1409-15.
114. Berry DA, Parmigiani G, Sánchez J y cols. Probability of carrying a mutation of breast-ovarian cancer gene BRCA1 based on family history. J Natl Cancer Inst 1997;89:227-38.
115. Johannesdottir G, Gudmundsson J, Berghorsson J y cols. High prevalence of the 999 del 5 mutation in Icelandic breast and ovarian cancer patients. Cancer Res 1996;56:3663-5.
116. Abeliovich D, Kaduri L, Lerer Y y cols. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of aerly-onset breast cancer patients among Ashkenazi women. Am J Hum Genet 1997;60:505-14.
117. Matsushima M, Kobayashi K, Emi M y cols. Mutation analysis of the BRCA1 gene in 76 Japanese ovarian cancer patients: Four germline mutation, but no evidence of somatic mutation. Hum Mol Genet 1995;4:1953-6.
118. Katagiri T, Emi M, Ito Y y cols. Mutations in the BRCA1 gene in Japanese breast cancer patients. Hum Genet 1996;7:334-9.
119. Miki Y, Katagiri T, Kasumi F y cols. Mutation analysis in the BRCA2 gene in primary breast cancers. Nat Genet 1996;13:245-7.
120. Easton DF, Ford D, Bishop DT y cols. Breast and ovarian cancer incidence in BRCA1 mutations carriers. Am J Hum Genet 1995;56:265-71.
121. Ford D, Easton DF, Bishop DT y cols. Risks of cancer in BRCA1 mutation carriers. Lancet 1994;343:692-5.
122. Ford D, Easton DF, Stratton M y cols. and Breast Cancer Linkage Consortion. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 1998;62:676-89.
123. Streuwing JP, Hartge P, Wacholder S y cols. Cancer risk with 185delAG and 5382insC mutations of BRCA1 and the 6174delT mutations of BRCA2 among Ashkenazi Jews. N Engl J Med 1997;336:1401-8.
124. Lynch HT, Marcus J, Watson P y cols. Distinctive clinicopathologic features of BRCA1-linkage hereditary breast cancer. Proc ASCO 1994;13:56.
125. Marcus JN, Watson P, Page DL y cols. Hereditary breast cancer. Pathobiology, prognosis and BRCA1 and BRCA2 gene linkage. Cancer 1996;77:697-709.
126. Lakhani SR, Sloane JP, Gusterson BA y cols. The pathology of familial breast cancer: Evidence for differences between breast cancers developing in carriers of BRCA1 mutations, BRCA2 mutations and sporadic cases. Lancet 1997;349:1505-10.
127. Lakhani SR, Jacquemier J, Sloane JP y cols. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst 1998;90:1138-45.
128. Eisinger F, Stoppa-Lyonnet D, Longy M y cols. Germline mutation at BRCA1 affects the histoprognostic grade in hereditary breast cancer. Cancer Res 1996;56:471-4.
129. Weber BL. Update on breast cancer susceptibility genes. Am Soc Clin Oncol. Educational book 1998:242-8.
130. Knudsen AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971;68:820.
131. Smith SA, Easton DF, Evans DG, Ponder BA. allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild type chromosome. Nat Genet 1992;2:128-31.
132. Holt JT, Thompson ME, Szabo C y cols. Growth retardation and tumor inhibition by BRCA1. Nat Genet 1996;12:298-302.
133. Tonin P, Weber B, Offit K y cols. Frequency of recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer families. Nat Med 1996;2:1179-83.
134. Streuwing JP, Abeliovich D, Peterz T y cols. The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 1995;11:198-200.
135. Roa BB, Boyd AA, Volcik K y cols. Ashkenazi Jewish population frequency for common mutations in BRCA1 and BRCA2. Nat Genet 1996;14:185-90.
136. Sobol H, Stoppa-Lyonnet D, Bressac-de-Paillerets B y cols. Truncation at conserved terminal regions of BRCA1 protein is associated with highly proliferating hereditary breast cancer. Cancer Res 1996;56:3126-9.
137. Merajver SD, Pham TM, Caduff RF y cols. Somatic mutations in the BRCA1 gene in sporadic ovarian tumors. Nat Genet 1995;9:439-43.
138. Hosking L, Trowsdale J, Nicolai H y cols. A somatic BRCA1 mutation in an ovarian tumor. Nat Genet 1995;9:343-4.
139. Takahashi H, Behbakht K, McGovern PE y cols. Mutation analysis of the BRCA1 gene in ovarian cancers. Cancer Res 1995; 55:2998-3002.
140. Neuhausen S, Gilewski T, Norton LJ y cols. Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer. Nat Genet 1996;13:126-8.
141. Lancaster JM, Wooster R, Mangion J y cols. BRCA2 mutations in primary breast and ovarian cancers. Nat Genet 1996;13:238-40.
142. Teng DHF, Bogden R, Mitchell J y cols. Low incidence of BRCA2 in breast carcinoma and others cancers. Nat Genet 1996;13:241-4.
143. Takahashi H, Chiu HC, Bandera CA y cols. Mutations of the BRCA2 gene in ovarian carcinomas. Cancer Res 1996;56:2738-41.
144. Thorlacius S, Streuwing JP, Hartge P y cols. Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 1998;352:1337-9.
145. Young JL, Perri CI, Asire AJ. National Cancer Institute Monograph 57, KY72 NIH Publication 1981, No 81-2330, Bethesda.
146. Malkin D, Li FP, Strong LC y cols. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas and other neoplasms. Science 1990;250:1233-8.
147. Li FP, Garber JE, Friend SH y cols. Recommendations on predictive testing for germ line p53 mutations among cancer-prone individuals. J Natl Cancer Inst 1992;84:1156-60.
148. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppresor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res 1994;54:4855-78.
149. Cornelis RS, Van Vliet M, Vos CBJ y cols. Evidence for a gene on 17p13.3 distal to p53, as a target for allele loss in breast tumors without p53 mutations. Cancer Res 1994;54:4200-6.
150. Marchetti A, Buttitta F, Pellegrini S y cols. p53 mutations and histological type of invasive breast carcinoma. Cancer Res 1993;53:4665-9.
151. Mazars R, Spinardi L, Bencheickh M y cols. p53 mutations occur in aggressive breast cancer. Cancer Res 1992;52:3918-29.
152. Brownstein MH, Wolf M, Bikowski JB. Cowden''s disease: A cutaneous marker of breast cancer. Cancer 1978;41:2393-8.
153. Starink TM. Cowden''s disease: Analysis of fourteen new cases. J Am Acad Dermatol 1984;11:1127-41.
154. Wood DA, Darling HH. A cancer family manifesting multiple occurrences of bilateral carcinoma of the breast. Cancer Res 1943;3: 509.
155. Nelen MRP, Padberg GW, Peeters y cols. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet 1996;13:114-6.
156. Tsou HC, Ping XL, Xie XX. The genetic basis of Cowden''s syndrome: Three novel mutations in PTEN/MMAC1/TEP1. Hum Genet 1998;102:467-73.
157. Spigelman AD, Murday V, Phillips RKS. Cancer and the Peutz-Jeghers syndrome. Gut 1989;30:1588-90.
158. Hizawa K, Iida M, Matsumoto T y cols. Cancer in the Peutz-Jeghers syndrome. Cancer 1993;72:2777-81.
159. Trau H, Schewach-Miller M, Fisher BK, Tsur H. Peutz-Jeghers syndrome and bilateral breast carcinoma. Cancer 1982;50:788-92.
160. Muir EG, Yates-Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum and larinx associated with keratoacathomata of the face. Br J Surg 1966;54:191-5.
161. Hall NR, Murday VA, Chapman P y cols. Genetic linkage in Muir-Torre syndrome to the same chromosomal region as cancer family syndrome. Eur J Cancer 1994;30A:180-2.
162. Anderson DE. An inherited form of large bowel cancer. Cancer 1980;45:1103-7.
163. Kinzler KW, Nilber MC, Su LK y cols. Identification of FAP locus genes from chromosome 5q21. Science 1991;253:661-5.
164. Groden J, Thliveris A, Samowitz W y cols. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589-600.
165. Papadopoulos N, Nicolaides N, Wei YF y cols. Mutation of mutL homolog in hereditary colon cancer. Science 1994;263:1625-9.
166. Bronner EC, Baker SM, Morrison PT y cols. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994;368:258-61.
167. Fishel R, Lescoe MK, Rao MRS y cols. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027-38.
168. Leach FS, Nicolaides N, Papadopoulos N y cols. Mutations of a mutS homolog in hereditary nonpolyposis colon cancer. Cell 1993;75:1215-25.
169. Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst 1986;77:89-92.
170. Swift M, Morrell D, Cromartie E. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet 1986;39:573-83.
171. Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991;325:1831-6.
172. Gatti RA, Berkel Y, Boder E y cols. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature 1998;336:577-80.
173. Ejima Y, Sasaki MS. Mutations of the ATM gene detected in Japanese ataxia telangiectasia patients: Posible preponderance of the two founder mutations 4612del165 and 7883del5. Hum Genet 1998;102:403-8.
174. Easton DF. Cancer risks in A-T heterozygotes. Int J Radiat Biol 1994;66:S177-S82.
175. Fraccaro M, Lindsten J, Ford CE y cols. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum Genet 1980;156:21.
176. Zackai EH, Emmanuel BS. Site-specific reciprocal translocation t(11;22)(q23;q11) in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet 1980;7:507-21.
177. Iselius L, Lindsten J, Aurias A y cols. The 11q;22q translocation: A collaborative study of 20 new cases and analysis of 110 families. Hum Genet 1983;64:343-55.
178. Lindblom A, Sandelin K, Iselius L y cols. Predisposition for breast cancer in carriers of constitutional translocation 11q;22q. Am J Hum Genet 1994;54:871-6.
179. American Society of Clinical Oncology. Statement of the American Society of Clinical Oncology: Genetic testing for cancer susceptibility. J Clin Oncol 1996;14:1730-6.
180. Biesecker B, Boehnke M, Calzone K y cols. Genetic counseling for families with inherited susceptibility to breast and ovarian cancer. JAMA 1993;269:1970-4.
181. Lynch HT, Watson P, Conway T y cols. DNA screening for breast-ovarian cancer susceptibility based on linked markers: A family study. Arch Intern Med 1993;153:1979-87.
182. Lerman C, Croyle R. Psychological illness in genetic testing for breast cancer susceptibility. Arch Intern Med 1994;154:609-16.
183. Lynch HT, Marcus JN, Watson P. Familial breast cancer, family cancer syndromes and predisposition to breast cancer. En: Bland KI, Copeland EM III, eds. The breast comprehensive management of benign and malignant disease. WB Saunders. Philadelphia 1991:262.
184. Fisher B, Costantino JP, Wickerham DL y cols. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998;90:1371-88.
185. Powles T, Eeles R, Ashely S y cols. Interin analysis of the incidence of breast cancer in the Royal Marsden Hospital Tamoxifen randomised chemoprevention trial. Lancet 1998;352:98-101.
186. Veronesi U, Maisonneuve P, Costa A y cols. Prevention of breast cancer with tamoxifen: Preliminary findings from the italian randomised trial among hysterectomised women. Lancet 1998;352: 93-7.
187. Eldar S, Meguid MM, Beatty JD. Cancer of the breast after prophylactic subcutaneous mastectomy. Am J Surg 1984;148:692-3.
188. Ziegler LD, Kroll SS. Primary breast cancer after prophylactic mastectomy. Am J Clin Oncol 1991;14:451-4.
189. Goldman LD, Goldwyn RM. Some anatomical considerations of subcutaneous mastectomy. Plast Reconstr Surg 1973;51:501-8.
190. Jarret JR. Prophylactic mastectomy. En Marsh JL (ed). Current therapy in plastic and reconstruction surgery. Deckker. Toronto, Canadá. 1989:64.
191. Pennisi VR y cols. Subcutaneous mastectomy data: A final statistical analysis of 1.500 patients. Aesthetic Plast Surg 1989;13:15.
192. Woods JE. Subcutaneous mastectomy. Current state of the art. Ann Plast Surg 1983;11:541-50.
193. Hartmann LC, Daniel J, Schaid PD y cols. Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 1999;340:77-84.
194. Stefanek ME, Helzlsoner KJ, Wilcox PM y cols. Predictors of and satisfaction with bilateral prophylactic mastectomy. Prev Med 1995;24:412-9.
195. Streuwing JP, Watson P, Easton DF y cols. Prophilactic oophorectomy in inherited breast-ovarian cancer families. J Natl Cancer Inst 1995;17:33-5.
196. Piver MS, Jishi MF, Tsukada Y y cols. Primary peritoneal carcinoma after Prophylactic oophorectomy in women with a family history of ovarian cancer. Cancer 1993;71:2751-5.