


El síndrome de Fraser es una enfermedad autosómica recesiva infrecuente. Su diagnóstico precisa al menos dos criterios mayores y uno menor, o bien un criterio mayor y cuatro menores (descritos por Thomas et al, en 1986). Presentamos el caso de una niña con dicho síndrome, nacida tras una gestación sin factores de riesgo, que no fue diagnosticado anteparto. Este caso presenta numerosos criterios diagnósticos (criptoftalmos, sindactilia, anomalías genitales...) y además algunas malformaciones muy infrecuentes en dicho síndrome como la ausencia de ovarios. Debemos sospechar este síndrome si la ecografía muestra oligoamnios, pulmones hiperecogénicos y voluminosos, agenesia renal y/o anomalías orbitarias.
The Fraser syndrome is an infrequent recessive autosomal disease. Two major criteria and one minor criterion or one major and at least four minor criteria are required for the diagnosis (provided by Thomas et al in 1986). We present the case of a newborn with Fraser syndrome, born after a no - risk pregnancy, that was not detected before the delivery. This case shows numerous diagnostic criteria (cryptophthalmos, syndactyly, abnormal genitalia...) and some very infrequent malformations as a part of this syndrome as the ovarian absence. We should think about this syndrome when the ecography shows oligohydramnios, hyperechogenic and voluminous lungs, renal agenesis and/or ocular malformations.
El síndrome descrito por Fraser en 19621 (Zehender2 en 1872 describió un caso de criptoftalmos asociado a malformaciones) es una enfermedad autosómica recesiva, más frecuente en fetos femeninos, con incidencia de 0,043/10.000 nacidos vivos y 1,1/10000 nacidos muertos3. La consanguinidad de los progenitores aparece en el 15-24,8% de los casos4–6. No existe evidencia de transmisión vertical. No existe método preventivo, salvo el consejo genético.
El criptoftalmos es la manifestación clínica más frecuente y consiste en la falta de formación de párpados, que deja la córnea y conjuntiva desprotegidas (de modo que estas estructuras pueden presentar una transformación dermoide). También puede cursar con alteraciones del segmento anterior y se pueden llegar a sustituir por tejido conectivo las estructuras del canal de Schlemm.
El diagnóstico precisa al menos 2 criterios mayores y uno menor, o bien un criterio mayor y cuatro menores (descritos por Thomas et al6 en 1986).
Caso clínicoLa paciente, secundigesta, de 31 años y 36+6 semanas de gestación (SG), fue remitida a la Unidad de Patología Obstétrica hospitalaria por cesárea anterior (por no progresión de parto) con evolución normal. No refería otros antecedentes de interés. Su pareja era un varón sano de 30 años, no consanguíneo. El control y el curso gestacional fueron normales. En las 38+6 SG se realizó una ecografía (ECO) básica en la que se detectó polihidramnios y presentación cefálica inestable. Dada la inmadurez cervical (Bishop 0), junto a hidramnios y signos de desproporción pélvico-cefálica (DPC), se optó por la realización de cesárea a las 39+6 SG. Nació un polimalformado, hembra, con un Apgar 7-9, 3.040g, con polihidramnios y líquido teñido. El puerperio transcurrió sin anomalías.
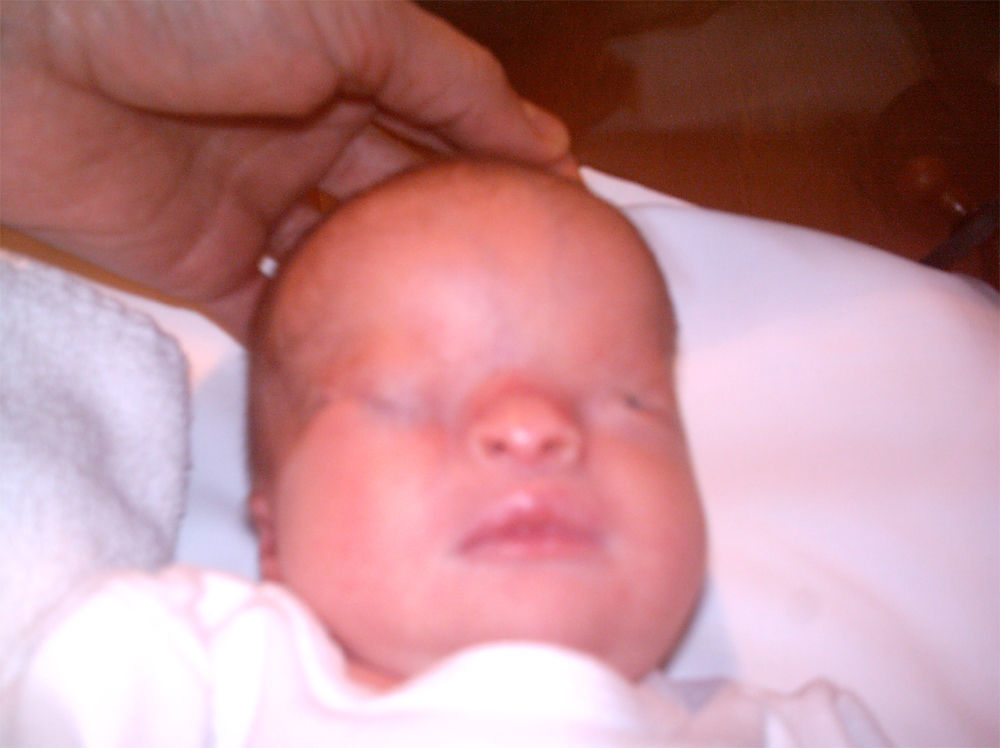
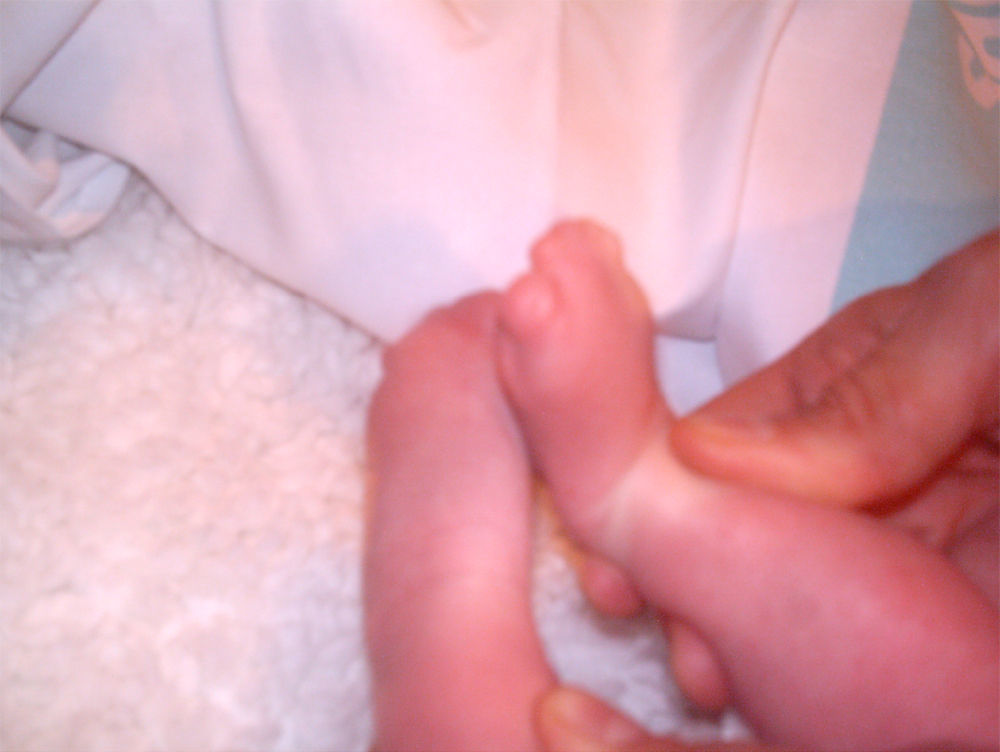
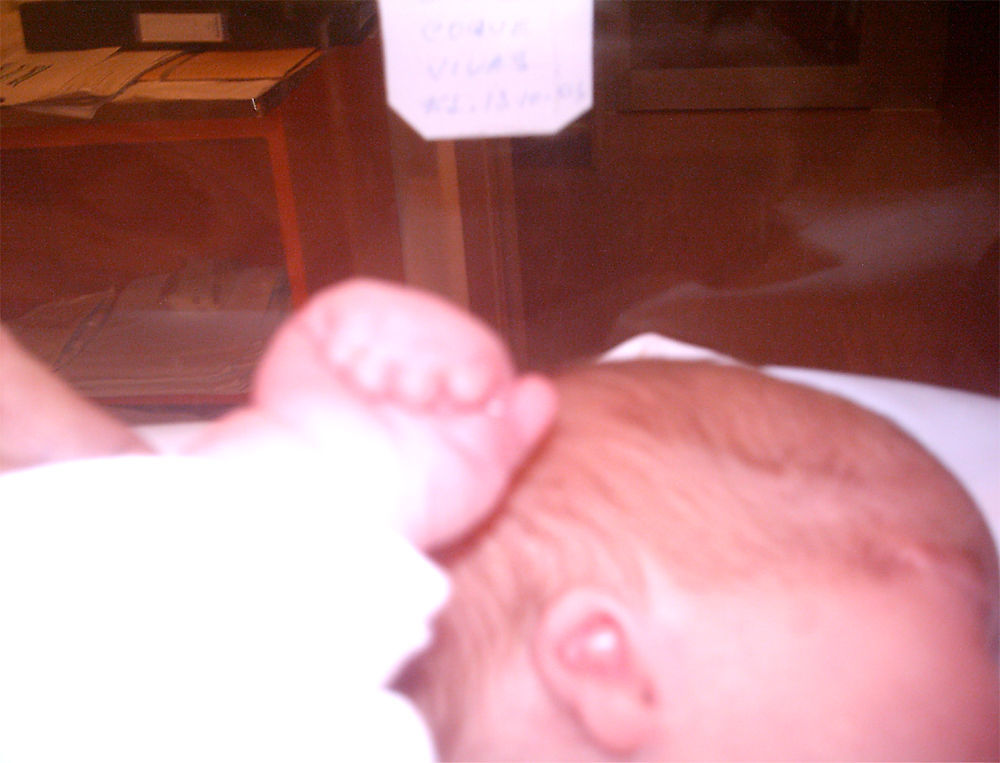
La recién nacida fue ingresada en la Unidad de Neonatología. En la exploración física presentaba dismorfia craneal, pabellones auriculares hipoplásicos y de implantación baja, anomalías oculares (déficit completo de hendidura palpebral e hipoplasia orbitaria en el ojo derecho, hendidura mínima y esbozo malformativo del globo ocular izquierdo), paladar ojival, hipoplasia nasal y secreciones abundantes en las vías respiratorias (figs. 1 y 3). En las extremidades presentaba sindactilia de manos y pies (figs. 2 y 3) Se observaban genitales externos de aspecto femenino con labios mayores y menores hipoplásicos, hipertrofia de clítoris e introito vaginal estenosado. No se identificaban ovarios, anomalía muy infrecuente. Se objetivaron peso y talla normales (3.040g, 51cm), ausencia de soplos cardíacos y abdomen normal a la palpación.



En las pruebas complementarias realizadas se observó cariotipo normal 46XX (igual que la mayoría de casos descritos), hipoplasia laríngea, estenosis subglótica e hipoplasia traqueal. La ECO transfontanelar fue normal. En la ECO abdominal se detectó agenesia renal unilateral (con un riñón presente de aspecto normal y función renal conservada) y no se identificaron ovarios. En la ECO ocular se identificaba globo izquierdo con cristalino y nervio óptico, y en la órbita derecha se observaba formación bicameral de paredes groseras y ausencia de nervio óptico y cristalino. El estudio cardiológico (electrocardiograma y ECO-cardiograma) fue normal.
Las analíticas generales eran normales (incluidas la urea y la creatinina), salvo fósforo (8,2mg/dl), LDH (332 U/l) y potasio (6,2mEq/l) elevados. Los análisis hormonales realizados mostraron cortisol basal y ACTH normales, 11-desoxicortisol 89,50ng/ml (elevado), estradiol 11pg/ml, LH 0,34 mU/ml, FSH 7,60mU/ml, testosterona 0,17ng/ml, androstendiona 2,80ng/ml y S-dehidroepiandrosterona 1,27μg/ml. La 17-hidroxiprogesterona (7,90ng/ml) y la prolactina (15,05ng/ml) estaban elevadas. El estudio tiroideo fue normal; GH e IGF-BP3 eran normales; la IGF-I estaba disminuida (< 10ng/ml). La actividad de la renina plasmática (28,23ng/ml/h) y la aldosterona (1.495pg/ml) estaban elevadas.
En el electroencefalograma se objetivó una disfunción eléctrica difusa con predominio derecho. En los potenciales evocados se obtuvieron respuestas retrasadas, con posible componente neurosensorial.
El grupo y Rh eran A positivo, con prueba de Coombs directa negativa. Se realizó aspirado nasofaríngeo y cultivo viral con resultado negativo. Se realizan serologías: toxoplasma inmunoglobulina G (IgG) (−), rubéola IgM (−), citomegalovirus (Fc) positivo 1/128, herpes virus (Fc) positivo 1/64, parvovirus B-19 199 (+) y parvovirus IgM (−).
Durante su estancia en neonatología, presentó anomalías en la succión de alimentos junto con dificultad respiratoria con secreciones en vías altas, motivados por alteraciones laríngeas y estenosis subglótica. Evolucionó favorablemente y fue dada de alta a los 22 días de vida. Los datos clínicos y exploratorios permitían realizar el diagnóstico definitivo de síndrome de Fraser.
Precisó 3 reingresos en los dos primeros meses de vida por insuficiencia respiratoria y cianosis. Posteriormente, presentó parada cardiorrespiratoria en su domicilio; tras su reanimación activa y el traslado inmediato en unidad de cuidados intensivos móvil al centro hospitalario, se produjo asistolia y la defunción de la recién nacida a los 73 días de vida.
DiscusiónEl caso descrito destaca fundamentalmente en dos aspectos. En primer lugar, cumple numerosos de los criterios diagnósticos para definirlo como síndrome de Fraser (tres mayores y cuatro menores); pocos casos se aproximan tanto a los criterios definitorios. En segundo lugar, se trata de un síndrome muy infrecuente (sólo existen 117 casos publicados5 desde que Thomas et al6 describieron los criterios diagnósticos hasta el año 2002; si tenemos en cuenta series previas al establecimiento de los criterios diagnósticos, hay descritos cerca de 400 casos en la literatura científica)7.
Los criterios mayores (y su frecuencia) son: criptoftalmos (93-88%: 27,4% unilateral y 53% bilateral), sindactilia (54-79%), anomalías genitales (criptorquidia, hipospadias, pene hipoplásico, clitoromegalia, atresia vaginal, útero bicorne o rudimentario, genitales externos ambiguos...) y hermanos afectados. Los menores son alteraciones de nariz, oídos, laringe, paladar, hernia umbilical, agenesia renal uni o bilateral, anomalías esqueléticas y retraso mental5,6,8.
Según Thomas et al6 y Koening and Spranger9, el criptoftalmos es la malformación más importante para el diagnóstico de este síndrome, pero no una condición necesaria para el diagnóstico, y abogan por el término «síndrome de Fraser» y no por «síndrome de criptoftalmos».
Con respecto a los hallazgos clínicos del caso descrito, la incidencia de paladar ojival en los casos de síndrome de Fraser es del 3,4% y la de hipoplasia nasal del 11,1%. La hipoplasia laríngea aparece en el 30,8% de casos y la estenosis subglótica e hipoplasia traqueal en el 8,5%. La hipertrofia de clítoris es la anomalía genital más frecuente en las mujeres con este síndrome (incidencia 36,8%) y sólo el 8% de casos presentan estenosis del introito vaginal. La ausencia de ovarios es muy infrecuente (tan sólo 2 casos de los 117 del estudio de Slavotinek tenían agenesia gonadal). La agenesia renal está presente en el 22,2% de casos. La mayoría de los casos descritos tienen cariotipo femenino normal, como sucede en nuestra paciente5.
En 2003, Mc Gregor et al10 identificaron el gen responsable de la enfermedad como el 4q21 o FRAS 1, que codifica una proteína de matriz extracelular. Sus mutaciones se traducen en la finalización temprana de la síntesis de la proteína que codifica dicho gen. También se conocen otras mutaciones causantes de la enfermedad en otros genes distintos del FRAS 1.
No existen estudios endocrinológicos en estos pacientes, a pesar de la alta frecuencia de anomalías genitales8. Las determinaciones realizadas en nuestro caso no tienen así referencias para realizar una comparación.
El diagnóstico antenatal es posible a partir de las 18 semanas, con alta sospecha en casos que asocien agenesia renal, anomalías orbitarias y aumento del volumen torácico. Debemos sospechar este síndrome si la ecografía muestra oligoamnios (en relación con los casos de agenesia renal bilateral/malformaciones renales severas) y pulmones hiperecogénicos y voluminosos (retención de líquido pulmonar fetal debido a estenosis laringo-traqueal; en estos casos podría aparecer polihidramnios, como en el caso descrito), que aparecen en el neonato como hiperplasia pulmonar11–13.
La supervivencia es variable y el pronóstico malo, por lo que el diagnóstico prenatal toma especial importancia. El 6% de casos son prematuros o abortos espontáneos, el 12% termina en interrupción voluntaria del embarazo y el 20% fallece en la primera semana de vida. El 13% sobrevive hasta 1 año de vida y otro 13% sobrevive entre 1 y 10 años5. Las causas determinantes de la defunción son la estenosis/atresia laríngea, insuficiencia respiratoria, agenesia renal bilateral o una combinación de malformaciones renales y respiratorias. Los pacientes afectados que sobrevivieron más de 10 años apenas tenían malformaciones mayores y dudosamente cumplían con los criterios diagnósticos que hemos descrito. En otras series, el 50% sobrevive 1 año o más (aunque el 25% eran mortinatos)14. Los casos que sobreviven presentan retraso mental grave7.
El caso no tenía antecedentes familiares conocidos de malformados, mientras que el 41% de los casos tiene historia familiar de criptoftalmos o hallazgos compatibles con síndrome de Fraser5.
No se objetivaron anomalías musculoesqueléticas (el 8-10% de los casos las presentan) ni gastrointestinales (7-12% de los casos). Aunque las malformaciones digestivas no son criterios diagnósticos, Slavotinek et al5 recomiendan que deberían considerarse, dada la frecuencia con que aparecen (atresia anal o anorrectal, estenosis anal, ano imperforado…).
El desarrollo posnatal fue deficiente (peso < P3 y talla P3-10), mientras que lo habitual es un crecimiento posnatal normal. Sólo el 18% de los casos de este síndrome presentan retraso de crecimiento5.
Este caso destaca además por presentar alguna malformación extremadamente infrecuente como parte del síndrome de Fraser, como paladar ojival (incidencia 3,4%), hipoplasia nasal (incidencia 11,1%) y ausencia de ovarios (tan sólo 2 casos de los 117 del estudio de Slavotinek et al5 tenían agenesia gonadal).