


Presentamos el caso de una paciente de 18 años que consultó por oligomenorrea, flujo menstrual maloliente y dolor pélvico tipo cólico. Ante la sospecha de alteración menstrual por defecto en el tracto de salida se realizó ecografía ginecológica y resonancia magnética con el resultado de útero doble, hemivagina obstruida o ciega derecha acompañado de agenesia renal derecha, síndrome Herlyn-Werner-Wunderlich. Tras el diagnóstico se realizó escisión del septo y drenaje de las colecciones que suprimen la sintomatología de la paciente mejorando la capacidad reproductiva.
We report the case of an 18-year-old woman who consulted for oligomenorrhea, chronic pelvic pain and malodorous vaginal discharge. Ultrasonography and magnetic resonance imaging demonstrated uterus didelphys with right-sided hematometrocolpos and absent right kidney (Herlyn-Werner- Wunderlich syndrome). The patient underwent resection of the septum and drainage of collections, with complete symptom resolution.
El síndrome de Herlyn-Werner-Wunderlich es una anomalía congénita poco frecuente, en la que coexiste la existencia de duplicación uterovaginal, con hemivagina obstruida o ciega y agenesia renal ipsilateral. Purslow, en 1922, fue el primer autor que presentó un caso con útero didelfo y hemivagina obstruida sin referencia a agenesia renal1, y ese mismo año Miller presentó un caso de útero didelfo con hemivagina obstruida y agenesia renal homolateral2. En 1971 a la asociación de agenesia renal y hemivagina ciega ipsilateral se la denominó síndrome de Herlyn-Werner y la asociación de aplasia renal unilateral junto con útero didelfo, hematocérvix y vagina simple se describió en 1976 por Wunderlich3.
Actualmente también se le conoce con el nombre de síndrome OHVIRA por sus siglas en inglés (uterine didelphys associated with Obstructed Hemivagina and Ipsilateral Renal Anomaly), término más amplio, ya que incluye otro tipo de anomalías renales aparte de la agenesia renal4.
Esta anomalía es consecuencia del no desarrollo o falta de fusión de los conductos de Müller. El origen embriológico común del aparato reproductivo y urinario explica la coexistencia de anomalías urinarias asociadas a malformaciones genitales en un porcentaje no despreciable de casos. Entre las alteraciones del tracto urinario asociadas con mayor frecuencia a anomalías del desarrollo de los conductos müllerianos se encuentran la agenesia renal, doble sistema colector, duplicación renal y riñón en herradura5.
Los síntomas se suelen presentar poco después de la menarquia, cuando la formación de hematocolpos deriva en dismenorrea y masa a nivel de Douglas, así como alteraciones en el patrón menstrual (oligomenorrea o amenorrea). Sin embargo, en un porcentaje elevado de casos los síntomas inespecíficos junto con un patrón menstrual normal hacen difícil su diagnóstico precoz.
Las complicaciones iniciales pueden manifestarse en forma de piohematocolpos, piosalpinx o incluso pelviperitonitis. A más largo plazo puede aparecer endometriosis, adherencias pélvicas y riesgo aumentado de infertilidad6,7.
Tradicionalmente, la técnica más apropiada para el diagnóstico y clasificación de las anomalías de los conductos de Müller ha sido la resonancia magnética, desempeñando también un papel importante la ecografía ginecológica2. Sin embargo, actualmente algunos autores consideran que el diagnóstico podría ser ecográfico mediante ecografía 3D transvaginal, recurriendo a la resonancia magnética en aquellos casos complejos o de difícil valoración8.
El tratamiento es quirúrgico conservador y consiste en la resección del tabique vaginal y marsupialización de la hemivagina ciega, así como el drenaje de las colecciones si las hubiera2.
El presente artículo tiene como objetivo describir un caso clínico que ilustra este proceso patológico, detallando el método diagnóstico y el tratamiento aplicados.
Descripción del casoPaciente de 18 años de edad que acudió a la consulta por presentar amenorrea secundaria (solo 4 reglas desde la menarquia), con última regla hacía 5 meses.
Se realizó una anamnesis y exploración en la que se objetivó: antecedentes familiares y personales sin interés, con menarquia a los 12 años. La paciente presentaba acné, hirsutismo y obesidad leve. A la exploración genital se visualizaron genitales externos normales, desarrollo puberal normal, no realizándose especuloscopia ni tacto vaginal por ausencia de relaciones sexuales.
En la ecografía abdominal realizada inicialmente en la consulta, con escasa repleción vesical, se objetivó un aparato genital interno normal.
Como pruebas complementarias se solicitó una analítica general y perfil hormonal que fue normal salvo un cociente LH/FSH algo aumentado.
Con los datos obtenidos se sospechó síndrome de ovario poliquístico y se pautó tratamiento con anticonceptivos orales combinados.
La paciente acudió a revisión; refería haber tomado el tratamiento con anticonceptivos un mes con posterior abandono, y presentar amenorrea secundaria, con flujo vaginal sanguinolento maloliente y dolor en el hipogastrio de tipo cólico durante los periodos de sangrado.
Se realizó una exploración física en la que se apreció vagina de aspecto normal, con sensación de masa lateral dolorosa.
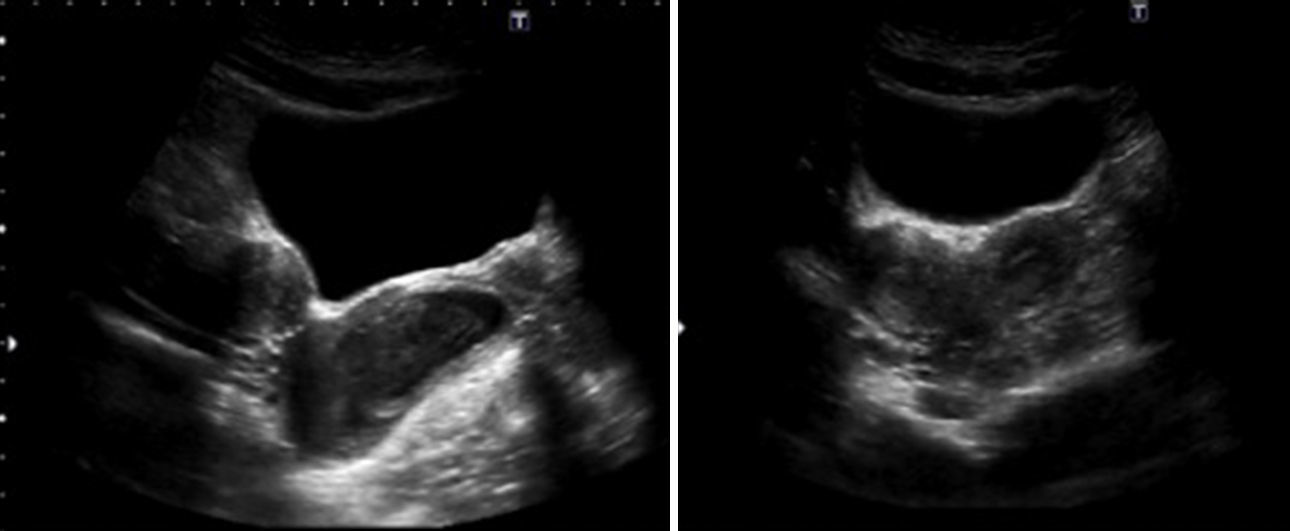
Ante la tríada de flujo maloliente, dolor de tipo cólico y amenorrea se sospechó una alteración en el tracto de salida, solicitándose una ecografía ginecológica en la que se visualizó útero doble, colección paravaginal derecha y vagina tabicada, sin visualizar imágenes anexiales patológicas (fig. 1).

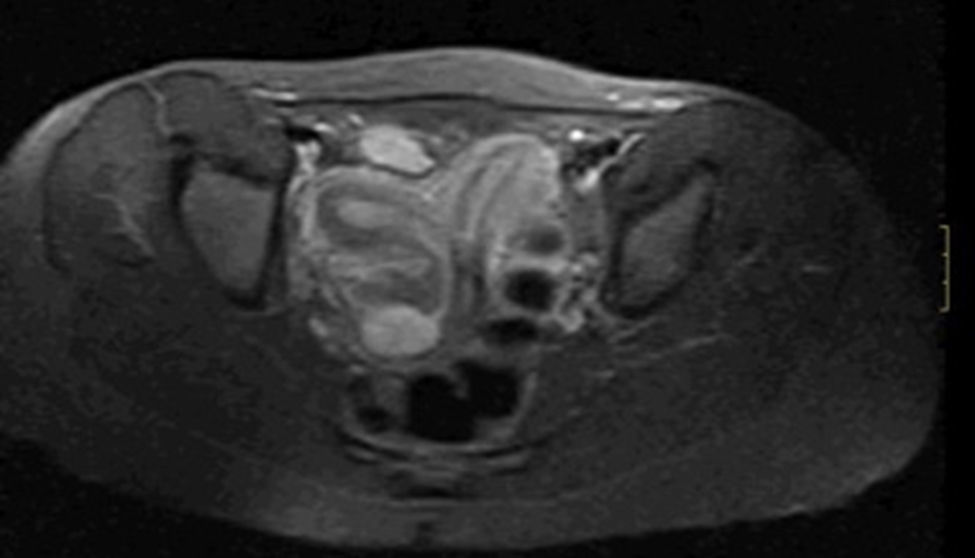
Ante los resultados de la ecografía se solicitó una resonancia magnética y una histerosalpingografía para determinar el tipo de malformación uterina y la dependencia de la colección paravaginal. En la resonancia magnética se objetivó un útero con morfología didelfo, colección lateralizada hacia la derecha, que se continúa con la cavidad cervical del hemiútero derecho y que es hipointensa en secuencias T1 e hiperintensa en secuencias T2, sugiriendo hidrocolpos a dicho nivel. Los ovarios eran normales (fig. 2).

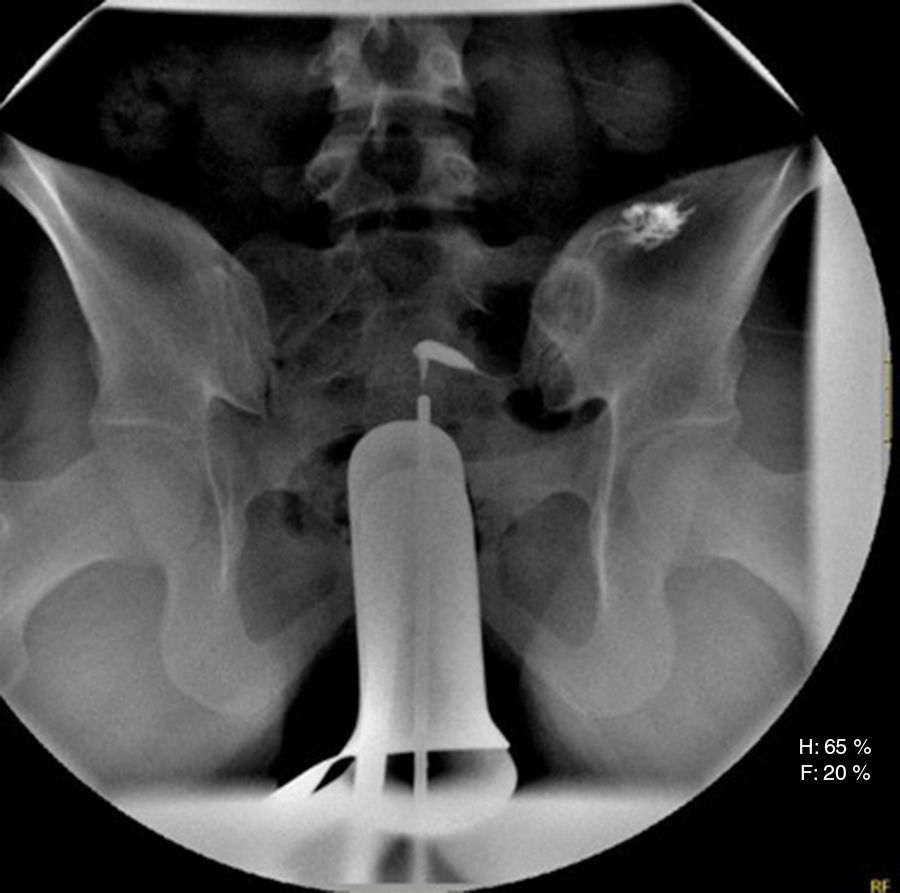
En la histerosalpingografía se visualizó hemiútero izquierdo permeable en el contexto de un útero didelfo y hemiútero derecho no visualizado debido a la presencia de tabique vaginal (fig. 3).

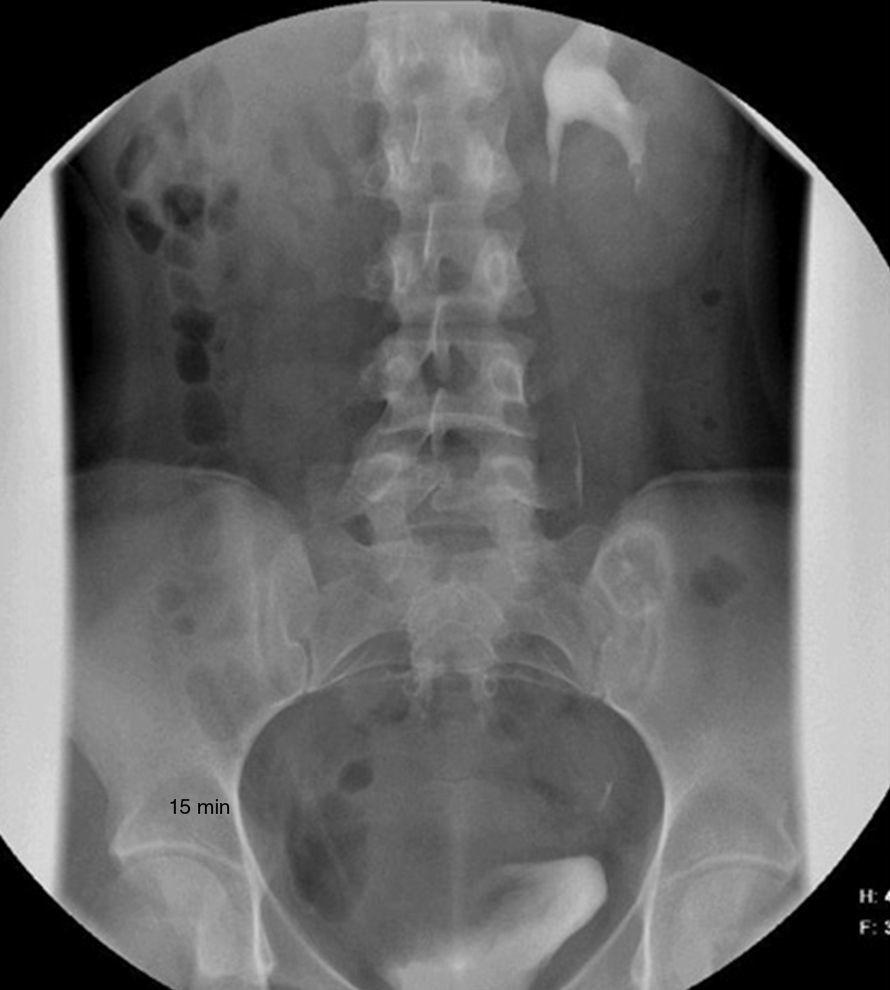
Dada la frecuente asociación entre anomalías renales y genitales se solicitó una urografía intravenosa en la que se observó la presencia de un riñón único izquierdo (fig. 4).

Nos encontramos por lo tanto con la asociación de duplicación uterovaginal con hemivagina obstruida o hemivagina ciega acompañada de agenesia renal ipsilateral (síndrome de Herlyn-Werner-Wunderlich).
Debido a la dificultad en las características exactas de la malformación por medio de la exploración y la resonancia se realizó una exploración vaginal bajo anestesia general y una laparoscopia diagnóstica, objetivando un hemiútero izquierdo permeable y en la pared derecha vaginal un cérvix rudimentario aumentado de tamaño, con drenaje de material sanguinolento a través de un orificio puntiforme, así como un útero doble con trompas y ovarios de características normales cada uno dependiente de su hemiútero.
El tratamiento consistió en la escisión del septo y drenaje de las colecciones. Actualmente la paciente se encuentra asintomática, con mejoría drástica del dolor tras la intervención y con ciclos menstruales regulares de entre 30-35 días. Continúa revisiones periódicas por el servicio de ginecología.
DiscusiónPara entender la patogenia del síndrome de Herlyn-Werner-Wunderlich es preciso repasar el origen embriológico del aparato genitourinario en la mujer. Los genitales internos y el tracto urinario derivan de 2 pares de estructuras que se desarrollan en ambos sexos: los conductos mesonéfricos o de Wolff y los conductos paramesonéfricos o de Müller. Los conductos de Wolff se comportan como inductores y hacen que los 2 conductos de Müller entren en contacto y se fusionen para formar el canal uterovaginal, que deriva en su parte proximal en las trompas de Falopio y en su parte distal en el útero, y los 2 tercios proximales de la vagina. Posteriormente se produce la reabsorción del tabique que los separa. El tercio inferior de la vagina procede del seno urogenital1. Como consecuencia del no desarrollo o del fallo de fusión de los conductos de Müller aparecen varios tipos de anomalías uterinas: hipoplasia, agenesia, útero bicorne, didelfo, útero septo o útero arcuato. Estas malformaciones han sido divididas en subclases por la Sociedad Americana de Medicina Reproductiva desde hace más de una década9 y es la clasificación de referencia.
Los conductos wolffianos, además de dar origen a los riñones, son elementos inductores de la fusión adecuada de los conductos müllerianos. Por este motivo, la anomalía en el desarrollo de la porción caudal de los conductos de Wolff puede ser la causa de la agenesia renal unilateral asociada. Del lado donde el conducto de Wolff está ausente, el conducto de Müller está desplazado lateralmente y no puede fusionarse con el conducto contralateral, resultando en útero didelfo. El conducto mülleriano contralateral da origen a una vagina mientras que el componente desplazado forma un saco ciego, la hemivagina obstruida o imperforada1,2. El introito vaginal no está comprometido, debido a su diferente origen, a partir del seno urogenital. Lo mismo ocurre con los ovarios, que derivan de las crestas gonadales, y con las trompas de Falopio, derivadas de la parte proximal del conducto de Müller. Por lo tanto, ante un diagnóstico de anomalía mülleriana se deben descartar anomalías urinarias asociadas, así como ante el hallazgo de agenesia o displasia renal en mujeres debemos descartar la existencia de malformaciones genitales asociadas1,3,10 con el fin de ofrecer un tratamiento precoz que evite la producción de hematocolpos, hematometra y menstruación retrógrada.
La etiología exacta del síndrome es desconocida. Se ha observado un patrón de herencia poligénico, además de posibles efectos ambientales como la exposición durante el desarrollo embrionario a radiaciones ionizantes, infecciones intrauterinas o tóxicos como el dietilbestrol7.
El septo que se forma de la unión de ambos conductos müllerianos para formar el canal uterovaginal degenera gracias a un proceso apoptótico inducido por el gen bcl2. La ausencia de este gen en las células hembras podría explicar parte de la etiología del útero septo en humanos7.
Se ha observado que estas anomalías se presentan de manera más frecuente en el lado derecho, sin existir una explicación clara11.
La prevalencia del síndrome es indeterminada, con cifras en la bibliografía que van desde 1/2.000 hasta 1/28.00012. Diversos estudios han intentado determinar la incidencia real de estas anomalías con resultados dispares, encontrando cifras que van del 0,1 al 3,8%. Sin embargo parece ser que se sitúa en torno al 2-3% en la población con problemas de fertilidad y del 0,3-1% en la población general8. Debido a su carácter raro el número de casos publicado es escaso6,7,13.
El modo de presentación más habitual en las series publicadas es la presencia de dolor pélvico recurrente, masa a nivel de Douglas (asociada a hematocolpos o hematómetra), flujo vaginal maloliente y alteraciones menstruales poco tiempo después de la menarquia. Estos síntomas se presentan debido a la formación de hematocolpos con la menstruación que deriva en dismenorrea1,14.
En contraste con otras anomalías del tracto genital femenino, como el himen imperforado, la atresia vaginal o el septo vaginal transverso que se presentan con amenorrea, el diagnóstico de este síndrome se puede demorar muchos meses porque en la mayoría de las ocasiones la menstruación es normal debido a que la menstruación a través del hemiútero no obstruido suele ser regular10. Esto, junto con la presencia en muchos casos de síntomas inespecíficos, hace difícil su diagnóstico en un primer momento, y cuando comienzan con dismenorrea son usualmente tratadas con antiinflamatorios o anticonceptivos orales, retrasando el tratamiento adecuado6. Es lo que ocurre en nuestro caso en una primera consulta: la presencia de menstruación inicial tras la menarquia con amenorrea secundaria y dismenorrea hizo sospechar el diagnóstico de síndrome de ovario poliquístico y fue con la aparición de flujo vaginal maloliente y masa pélvica cuando se pensó en la existencia de una anomalía del tracto de salida como causa de la amenorrea.
Como hemos comentado el diagnóstico suele realizarse tras la menarquia. Sin embargo, se han descrito casos de aparición en el recién nacido como consecuencia de la influencia estrogénica materna15, así como demora hasta la edad adulta por la presencia de complicaciones derivadas de la presencia de hematocolpos y hematómetra que han pasado desapercibidos produciendo piocolpos como manifestación inicial14. En algunas ocasiones el diagnóstico se ha encontrado durante la gestación, por complicaciones como aborto espontáneo o parto pretérmino3,11,13. El pequeño tamaño del útero en la infancia hace casi imposible el diagnóstico en este grupo de edad.
Por lo tanto, las causas de retraso en el diagnóstico son la presencia de menstruación irregular a través del hemiútero no obstruido, el carácter raro de la enfermedad y el tratamiento de la dismenorrea con analgésicos o anticonceptivos que pueden mejorar los síntomas y enmascarar el cuadro10.
Los métodos diagnósticos más adecuados son la resonancia magnética y la ecografía 3D transvaginal8. El diagnóstico correcto se basa en la obtención de un corte coronal que permita valorar la morfología de la cavidad y su relación con el contorno externo. La ecografía 3D transvaginal permite esta valoración de manera bien estandarizada. Los grupos de mayor experiencia presentan una concordancia muy alta entre la valoración 3D transvaginal y la resonancia, con alta sensibilidad y especificidad8. Sin embargo, la falta de experiencia o disponibilidad de esta técnica, así como la posibilidad de diagnóstico urológico adicional han hecho que la resonancia magnética siga siendo la técnica de primera elección.
Frecuentemente se han utilizado también la histerosalpingografía y la cistografía retrógrada para detectar defectos de depleción a nivel urogenital, debido a su bajo coste. La laparoscopia no es una técnica de primera elección, pero puede ayudar cuando las imágenes radiológicas no son concluyentes o cuando las técnicas de imagen no están disponibles. Además, puede ser terapéutica en casos seleccionados: drenaje de hematocolpos, septoplastia o marsupialización de la hemivagina ciega6.
Sin un tratamiento adecuado la historia natural de la enfermedad deriva en una serie de complicaciones a corto y largo plazo. A corto plazo se produce piohematocolpos, piosalpinx, hematómetra o piometra5. A largo plazo se puede producir endometriosis debido a menstruación retrógrada, infección de las colecciones con la aparición incluso de pelviperitonitis, adherencias pélvicas y riesgo aumentado de abortos de repetición.
En la disminución de la fertilidad intervienen diversos factores: la hipovascularización del septo que impediría una correcta implantación, la constricción de la cavidad uterina, la posible incompetencia cervical y la pérdida de masa muscular miometrial7. El hecho de que una paciente presente una malformación mülleriana no conlleva necesariamente problemas de infertilidad. En algunas series publicadas se encuentra un 67% de embarazos a término sin complicaciones, un 23% de abortos y un 15% de partos pretérmino6.
En cuanto al tratamiento la opción más adecuada es quirúrgica conservadora mediante técnicas sencillas que consisten en la escisión del septo, marsupialización de la hemivagina ciega y drenaje de las colecciones3,5,14. El tratamiento suprime el dolor y mejora la capacidad reproductiva, así como evita las complicaciones tardías (endometriosis, síndrome adherencial y colecciones). Tras la cirugía la cavidad uterina recupera su tamaño normal en aproximadamente 3 meses y las tasas de obstrucción recurrente son muy bajas.
Actualmente no se considera la hemihisterectomía como el tratamiento de primera elección, porque se ha encontrado una tasa similar de gestación en ambos hemiúteros por igual11.
Se puede considerar la utilización de anticonceptivos orales combinados previa a la cirugía para prevenir la acumulación de hematocolpos y mejorar la sintomatología.
En cuanto a la malformación renal se debe evaluar de manera periódica la función renal, así como se debe hacer hincapié en la prevención de las infecciones urinarias, al tratarse de pacientes con un solo riñón.
ConclusiónEl síndrome de Herlyn-Werner-Wunderlich es una anomalía de difícil diagnóstico, principalmente por su carácter raro. Las pruebas diagnósticas principales son la ecografía transvaginal 3D y la resonancia magnética. El diagnóstico temprano es fundamental para mejorar los síntomas y preservar la función sexual y reproductiva. El tratamiento es sencillo y consiste en la escisión del septo, drenaje de colecciones y marsupialización de vagina ciega.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.