






Entre la lateralidad habitual (situs solitus) y la imagen completa en espejo (situs inversus) se encuentra el situs ambiguo o heterotaxia. Sus dos modalidades principales son el isomerismo izquierdo (con poliesplenia) o el derecho (con asplenia). La heterotaxia implica alteraciones en la movilidad ciliar que dificultan la migración de órganos embrionarios. Presenta malposición de órganos toracoabdominales, cardiopatías complejas y otras malformaciones. Presentamos un caso de diagnóstico ecográfico prenatal de isomerismo izquierdo, hígado a la izquierda y asplenia, asociado a cardiopatía e interrupción de la vena cava inferior con continuidad de la ácigos. La lateralidad de los órganos fetales debe ser parte del examen ultrasonográfico rutinario, por las frecuentes malformaciones asociadas a la heterotaxia.
There is a spectrum of heterotaxic syndromes between normal organ distribution (situs solitus) and congenital conditions in which major organs are mirrored from their normal position (situs inversus). The two main modalities are left isomerism (with polysplenia) and right isomerism (with asplenia). Heterotaxic defects involve ciliary dyskinesia, hampering migration of embryonic organs and leading to malposition of thoracic and abdominal organs, complex cardiac defects, and other malformations. We present a case of prenatal echographic diagnosis of levocardia, with left-sided liver, asplenia, congenital heart disease, and interruption of the inferior vena cava with azygos continuation. Because of the malformations frequently associated with heterotaxy, the position of fetal organs should form part of routine ultrasonographic examination.
Durante el desarrollo embrionario temprano, aproximadamente entre la quinta y la sexta semanas, se produce una asimetría derecha-izquierda fisiológica en los órganos toracoabdominales. Esta distribución de los órganos o situs puede presentar diversa variabilidad, con un amplio abanico de posibilidades: el situs solitus, que es lo que conocemos como lateralidad habitual; el situs inversus, en el que la distribución de los órganos presenta una imagen completa en espejo a la distribución habitual, y el situs ambiguus, también denominado situs inversus parcial, heterotaxia o isomerismo1. En este último, los patrones de distribución son muy variados y se presenta asociado a isomerismo y múltiples malformaciones en órganos torácicos y abdominales.
Su frecuencia es de 1/10.000 recién nacidos2,3.
El síndrome de heterotaxia tiene un pronóstico variable que depende de las malformaciones asociadas que presenta, fundamentalmente las cardiopatías, y ellas son las que condicionan, a su vez, el manejo perinatal. El caso que describimos presenta isomerismo izquierdo con interrupción de la vena cava inferior y continuidad en la ácigos, que es más frecuente en los casos de isomerismo derecho.
CASO CLINICOPaciente secundigesta de 30 años, sin antecedentes personales ni familiares de interés, G3PN1A1, con amenorrea de 32 semanas, que acudió a nuestro centro con diagnóstico ecográfico de estómago en posición derecha y defecto en el tabique interauricular. Las ecografías previas que aportaba eran acordes. Se realizó una ecografía en la que se objetivó una biometría fetal acorde a 31 semanas, placenta posterior de grado II y líquido amniótico normal. En el estudio anatómico fetal se observó en el abdomen una cámara gástrica a la derecha y el hígado a la izquierda; no se observó el bazo y se objetivó una interrupción de la vena cava inferior con continuidad en la ácigos. El corazón en levocardia y levoápex, con ausencia del tabique interauricular (canal auriculoventricular incompleto tipo aurícula única), pero ambos ventrículos eran proporcionados y los grandes vasos estaban normorrelacionados. Asociaba una hendidura mitral sin signos de insuficiencia cardíaca ni de insuficiencia de las válvulas auriculoventriculares significativa (figs. 1-5).
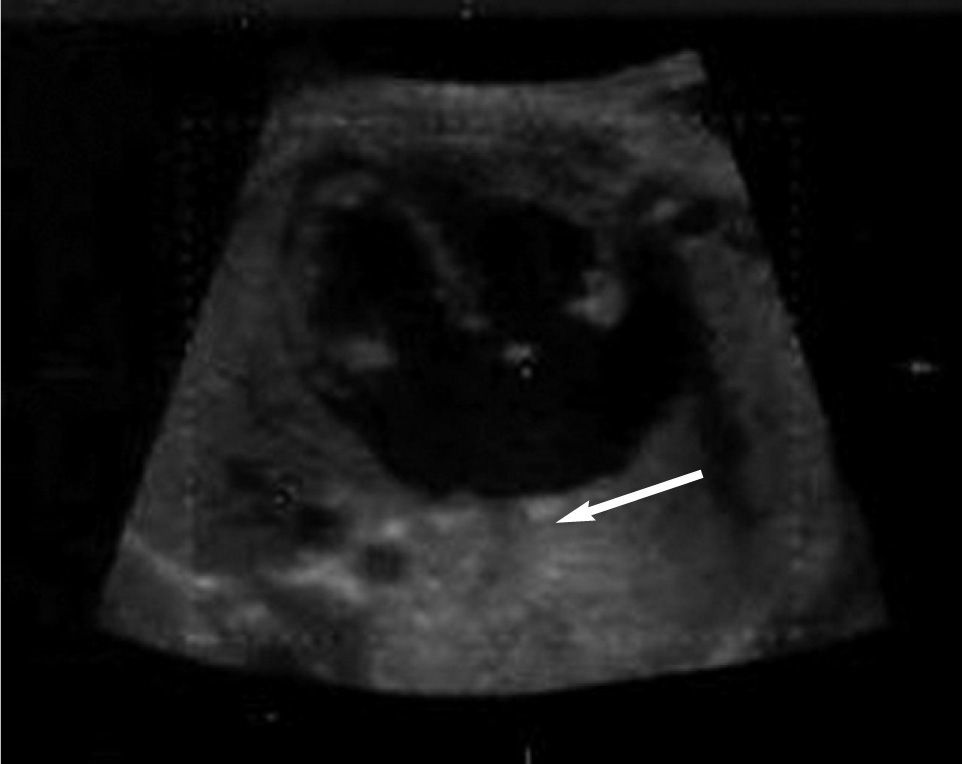
 es la estructura de referencia situada a la izquierda. El hígado (H) se encuentra a la izquierda, el estómago (E) a la derecha y el vaso a la derecha de la aorta y posterior es la vena ácigos (Az). Se referencia la columna (C), lado izquierdo (I) y derecho (D).")
Corte transversal a nivel del abdomen fetal. La aorta (Ao) es la estructura de referencia situada a la izquierda. El hígado (H) se encuentra a la izquierda, el estómago (E) a la derecha y el vaso a la derecha de la aorta y posterior es la vena ácigos (Az). Se referencia la columna (C), lado izquierdo (I) y derecho (D).
 son paralelos en la parte posterior del abdomen.")

. En este caso, ambas válvulas auriculoventriculares se encuentran abiertas.")

Se realizó una funiculocentesis para el diagnóstico citogenético, que informó de un cariotipo 46XX normal. Además realizamos una resonancia magnética en la que se confirmó la presencia de hígado y vesícula biliar centrales, cámara gástrica opuesta a silueta cardíaca, cámara auricular única y sendos ventrículos. Informaba de posible asplenia sin llegar a su confirmación.
Se realizó seguimiento ecográfico semanal, sin cambios significativos y con crecimiento fetal adecuado. El comienzo del parto fue espontáneo en la semana 39; nació una mujer de 3.020g, con una puntuación en la prueba de Apgar de 9 y 10 al min y a los 5min, respectivamente, pH de cordón 7,24 y 7,29, y que precisó reanimación neonatal de tipo I. Se ingresó a la recién nacida en neonatología donde se confirmó el diagnóstico prenatal y, al no presentar signos de insuficiencia cardíaca, fue dada de alta al quinto día posnatal. Se han realizado controles periódicos de la niña en las consultas de cardiología pediátrica, pero hasta el momento no aparecieron signos de insuficiencia cardíaca y no precisó tratamiento médico. La corrección quirúrgica del defecto cardíaco debe realizarse aproximadamente entre los 2 y 3 años de vida, si la clínica de la paciente lo permite.
DISCUSIÓNEl síndrome de heterotaxia es el resultado de un defecto en la organogénesis precoz que se produce en la semana quinta a sexta de gestación y presenta malposición de órganos toracoabdominales y vasos, cardiopatías complejas y otras malformaciones asociadas.
Su fisiopatología viene determinada por alteraciones en la motilidad ciliar, que producen dificultad en la migración de los órganos embrionarios, ya que los cilios son las organelas implicadas en las migraciones celulares y en la determinación de la lateralidad. Al analizar las mutaciones de los cilios se ha encontrado que las proteínas motrices moleculares cinesina II y dineína son fundamentales para esta función4. A partir de experimentos realizados en roedores, se han descrito cilios especiales, los monocilios, que se localizan en la región del nodo en la etapa de gastrulación, donde realizan movimientos hacia la izquierda, moviendo el líquido extraembrionario que rodea el nodo e iniciando así la asimetría derecha-izquierda5. De esta manera, cuando existe una anormalidad en la función ciliar, se puede alterar la lateralidad. La disfunción ciliar más frecuente es el síndrome de discinesia ciliar primaria6,7, una afección autosómica recesiva que se caracteriza clínicamente por distrés respiratorio neonatal, bronquiectasias, sinusitis y rinitis de inicio temprano e infertilidad. Los portadores de este síndrome tienen un 50% de situs solitus y 50% de situs inversus; este último tipo es el clásico síndrome de Kartagener. Por otro lado, el 20-25% de las personas con situs inversus son portadoras de discinesia ciliar primaria.
Existen evidencias de casos familiares con expresiones dominantes y recesivas; además, se ha observado, en parientes cercanos de estas personas, una mayor frecuencia de otras malformaciones «aisladas», lo que sugiere que estos defectos genéticos podrían estar implicados en la aparición de malformaciones esporádicas que no se asocian de una forma tan obvia a defectos de lateralidad, por ejemplo el síndrome de costillas cortas, la polidactilia, las displasias renal, hepática y pancreática, la agnatia y la holoprosencefalia8 Otro ejemplo de síndrome relacionado es el síndrome de Johanson-Blizzard, que consiste en la restricción de crecimiento intrauterino, situs inversus total, malformaciones cardíacas y gastrointestinales, y defectos del cuero cabelludo.
El bazo casi siempre se encuentra afectado en este síndrome, y su asociación con cardiopatías aparece en el 90% de los casos con asplenia, en el 70% de los casos con poliesplenia y en el 3% de los casos con situs inversus total. Las anomalías cardíacas más frecuentes son: transposición de grandes arterias, comunicación interauricular, comunicación interventricular, retorno venoso anómalo, aurículas isoméricas, canal auriculoventricular y bloqueo auriculoventricular9,10.
Clásicamente, Rose et al10 clasificaron el síndrome de heterotaxia en función de las alteraciones esplénicas que presentara. Según esto, pueden dividirse en casos asociados a asplenia, en los que es más frecuente la asociación con isomerismo derecho (estómago a la derecha, dextrocardia, aorta y cava inferior al mismo lado de la columna), cardiopatías graves, drenaje pulmonar anómalo y daño inmunitario importarte debido a la ausencia de bazo. Por todo ello, y fundamentalmente por las cardiopatías, estos casos están asociados a peor pronóstico, con elevada mortalidad en el primer año de vida. Por otro lado, están los casos asociados a poliesplenia, con mayor frecuencia de isomerismo izquierdo (transposición hepática, ausencia de vesícula biliar y malrotación intestinal) y que presentan múltiples bazos pequeños a ambos lados del abdomen del feto y cardiopatías menos graves. Aunque el pronóstico es algo mejor, la mortalidad también es elevada. No obstante, se considera que para un correcto conocimiento de cada caso de síndrome de heterotaxia es más útil describir con precisión la anatomía específica de cada paciente11,12.
El diagnóstico de este síndrome se centra en el estudio ecográfico, la resonancia magnética y el estudio citogenético. La ecografía es clave, ya que el estudio de la lateralidad debe formar parte del estudio ecográfico rutinario. Una vez diagnosticada la heterotaxia se debe realizar una ecocardiografía, debido a la alta frecuencia de malformaciones cardíacas asociadas, y una exploración abdominal cuidadosa, prestando especial atención a la posición del drenaje venoso (ácigos) que, en el caso presentado, aparecía afectado, la aorta, la presencia de vesícula biliar, malrotaciones y dilataciones intestinales, y presencia y número de bazos. La interrupción de la vena cava inferior con continuidad en la ácigos puede visualizarse mediante ultrasonidos: la vena ácigos asciende paralela y a la derecha de la aorta descendente y aparece más posterior de lo que habitualmente lo hace la vena cava, que mantiene una angulación de aproximadamente 45o respecto a la aorta13.
La resonancia magnética ayuda a confirmar el diagnóstico de poliesplenia y las posibles malrotaciones intestinales. Además, una de las posibles complicaciones de este síndrome es la obstrucción intestinal y la resonancia es útil para valorar la presencia de asas dilatadas y su posible localización14. El cariotipo fetal debe solicitarse siempre debido a que este síndrome se ha descrito asociado a alteraciones cromosómicas tipo monosomías, trisomías y translocaciones12,15.
Como conclusiones, debemos recordar que la lateralidad debe formar parte del examen ecográfico prenatal habitual. Una vez diagnosticada la heterotaxia, y debido a la alta frecuencia de malformaciones asociadas, debemos realizar un estudio minucioso cardíaco y abdominal. Hoy día, la resonancia magnética es un complemento para completar el diagnóstico y valorar las posibles complicaciones asociadas, como la obstrucción intestinal.